文献:首次人体试验的设计和行为注意事项
付永超 / 2019-11-25
首次人体试验的设计和行为注意事项
Jie Shen Brandon Swift Richard Mamelok Samuel Pine John Sinclair Mayssa Attar
First published: 26 July 2018 https://doi.org/10.1111/cts.12582
摘要:
将基础科学发现转化为治疗应用的转化科学的里程碑式一步是将候选药物从临床前研究开发到最初的人体测试。“首次人体(FIH)“试验是开发新的有前途的候选药物的链接,主要是为了确定进一步临床开发的安全剂量范围。跨职能协作对于确保高效,成功的"首次人体(FIH)“试验至关重要。本出版物的目的是作为针对小分子和生物药物候选者进行"首次人体(FIH)“试验的指南,主题涵盖法规要求,临床前安全性测试,研究设计注意事项,安全监控,生物标志物评估和全球注意事项。重点放在"首次人体(FIH)“试验设计的注意事项上,包括起始剂量选择,研究规模和群体,剂量递增方案以及适应性设计的实施。根据"欧洲药品管理局(EMA)“关于"首次人体(FIH)“试验指南的最新修订版,以提高安全性和降低风险,我们还讨论了该指南中引入的影响"首次人体(FIH)“试验设计的新措施。
成功开展"首次人体(FIH, First-in-human)“试验需要申办方在许多学科之间进行透彻的考虑和计划,以完成任务。这些学科需要无缝整合。从药理学的角度来看,体外和体内的动物数据均有助于提供有关候选"药物效力(potency )“和"药理学(pharmacologic )“特征的信息,以确认拟议临床适应症的相关性和潜力。临床前"药物代谢动力学(PK, pharmacokinetic)“数据可提供所需的"暴露-反应(exposure-response)“曲线,以评估"首次人体(FIH)“研究有效且有益的治疗相关剂量范围。体外代谢和"药物相互作用DDI)“研究表明,有必要在开发计划中尽早评估"药物相互作用(DDI)“风险。不仅需要对候选药物进行毒理学评估,以符合法规要求,而且理想情况下还可以使您了解靶标和靶标药理学,从而可以最大程度地降低对人类的转化风险。
临床药理学家通常是"首次人体(FIH)“研究的“所有者(owners)”,他们应确保研究设计考虑到有关候选药物的所有临床前"学问(Learnings)"。此外,“首次人体(FIH)“研究也需要被视为通向未来临床开发的“桥梁(bridges)”,因此,跨职能地开展工作以吸引关键利益相关者,才能最好地进行研究设计和实施。考虑到未满足的医疗需求和竞争格局,相关治疗领域的临床开发科学家应就研究设计要素提供输入,包括具体的疾病考虑因素和对"疗效(efficacy)“信号的任何潜在探索**。**制剂科学家提供输入以在中试和后期开发/商业化剂型之间取得平衡,以获得早期的相对生物利用度信息。安全医生应精心制定安全监控计划,这是任何"首次人体(FIH)“试验的关键组成部分。临床操作同事是关键的合作伙伴,可帮助进行"研究中心选择(study site selection)",启动活动,“机构审查委员会(IRB, institutional review board)”/“伦理委员会(EC, ethics committee)“批准,“合同研究组织(CRO, contract research organization)“合作以及研究监督,以确保研究的顺利进行和质量。主要利益相关者及其主要职责在图 1。
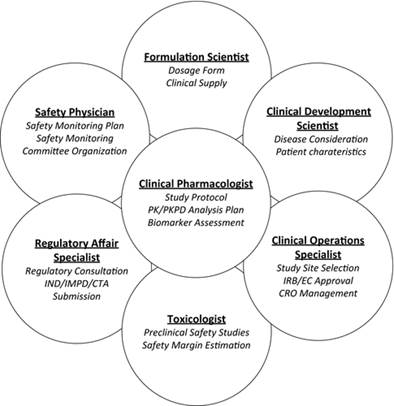
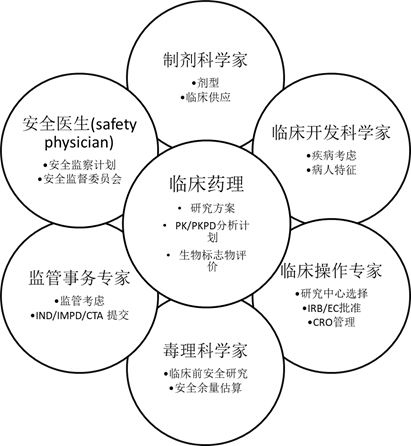
图一,关键利益相关者及其在计划"首次人体(FIH)“研究中的主要职责。 CTA,临床试验申请,clinical trial application; CRO,合同研究组织,contract research organization; EC,伦理委员会,ethics committee; IMPD,药品研究档案,Investigational Medicinal Product Dossier; IND,试验性新药,investigational new drug; IRB,机构审查委员会,institutional review board; PK,药物代谢动力学,pharmacokinetic; PKPD,药物代谢药物效应动力学,pharmacokinetic pharmacodynamic。
在进行"首次人体(FIH)“研究之前,始终要先在进行研究的国家/地区向卫生当局提交监管文件(例如,美国的研究性新药(IND)申请,欧洲的研究性药物产品档案等)。尽管无论是"美国食品和药物管理局(FDA)“和"欧洲药品管理局(EMA)“已经就如何开展"首次人体(FIH)“研究提供了有用的指南文件,但在其中不可能找到复杂药物开发项目中存在的所有问题的答案,特别是如果这些问题是治疗适应证特有的情况下。在某些情况下,申办方可能会从提交申请前的与监管部门的互动中受益,以将所学习到的知识应用"首次人体(FIH)“研究设计中,这不仅可以最大程度地降低由于安全性问题而导致的"临床暂停(clinical hold)“风险,而且还可以确保在追求早期"疗效(efficacy)“或"生物标志物(biomarkers)“探索时的投资回报。
认识到对"人类潜在的治疗媒介(potential therapeutic agents in humans)“进行初步试验的复杂性和多学科性质,本教程旨在对上述关键要素提供一些见解(Insights),以帮助申办方在"首次人体(FIH)“试验的设计和执行中更加成功。鉴于细胞和基因疗法以及疫苗在表征和监管预期方面存在显着差异,因此针对这些方法的"首次人体(FIH)“试验不在本教程的讨论范围之内。
临床前安全性测试Preclinical safety testing
为支持"首次人体(FIH)“试验而制定临床前安全计划的参考文献和指导文件有很多。特别是在"国际人用药品注册技术协调会(ICH, International Conference on Harmonization)“M3(R2)《支持药物进行临床试验和上市的非临床安全性研究指导原则》中就"国际人用药品注册技术协调会(ICH)“监管部门对进行"首次人体(FIH)“试验安全性评估计划的期望提出了建议。“国际人用药品注册技术协调会(ICH)” S6(R1)《生物制品的临床前安全性评价》提供了有关生物技术衍生药物(生物制剂)特定要求的其他信息,“国际人用药品注册技术协调会(ICH)” S9《抗肿瘤药物非临床评价指导原则问答》提供了有关抗癌药物非临床评估的指南。在制定适合特定药物开发项目(即"疗法(modality)“和"适应症(indication)")需求的临床前安全性测试策略时,牢记这些监管预期的背景是有帮助的。
临床前安全性测试有几个主要目标:i)识别器官毒性,ii)毒性与药物暴露的关系,iii)确定靶标和脱靶效应,iv)与人类的潜在相关性,以及v)鉴定/确认安全性生物标志物,以用于临床监测。实现这些目标的方法将取决于一些因素。首先,“治疗(therapeutic)“媒介的类型(例如,小分子或生物制剂)将决定进行"首次人体(FIH)“临床试验必不可少的研究以及监管机构预期的研究(表 1)。其次,需要"治疗(therapeutic)“的适应症(例如心血管,中枢神经系统(CNS),肿瘤学和罕见/孤儿疾病)将决定是否需要进行额外的评估和考虑,以更好地了解靶器官中与剂量相关的"疗效(efficacy)“和"毒性(toxicity)",以评估"安全性冗余量(safety margin)"。最后,必须考虑"首次人体(FIH)“试验和后续研究的范围和设计(例如治疗持续时间),以便设计适当的关键临床前研究,这样就既可以进行计划中的临床研究,又可以充分告知目标患者或健康的志愿者他们潜在的安全风险。
表1 ICH recommended preclinical studies enabling FIH trials
| Study type | Small molecules | Large moleculesa | GLP compliance Requirement |
|---|---|---|---|
| Pharmacodynamics | No | ||
| In vitro (MOA) | X | X | |
| In vivo (MOA and therapeutic effect) | X | X | |
| Safety pharmacology ICH S7A and S7B) | Yes | ||
| In vitro (concentration‐effect relationship) | X | X | |
| In vivo (dose‐response for CNS, CV, respiratory effects) | X | X | |
| Pharmacokinetics (ICH M3(R2)) | |||
| In vitro metabolism (across species microsomal metabolism) | X | NA | No |
| In vitro plasma protein binding | X | NA | No |
| Toxicokinetics from repeat dose GLP toxicity studies (ICH S3A) | X | X | Yes |
| Genotoxicity battery (ICH S2(R1)) | Yes | ||
| In vitro Ames test | X | *b | |
| In vitro and/or in vivo mammalian cell chromosomal damage evaluation | X | *b | |
| Single‐dose / dose range finding | No and Yesc | ||
| Rodent single‐dose (could be MTD study) | X | NA | |
| Nonrodent single‐dose (could be MTD study) | X ^e | X ^f | |
| Repeat dose toxicity ^d(ICH M3(R2)) | Yes | ||
| Rodent multidose | X | Optionalf | |
| Nonrodent multidose | X ^e | X ^f | |
| Other studies | No | ||
| Immunotoxicity (ICH S8) | X | X | |
| Photosafety (ICH S10) | X | X | |
| Abuse liability | X | X |
表1 ICH建议开展FIH试验的临床前研究
| 研究类型 | 小分子 | 大分子 ^a | GLP合规要求 |
|---|---|---|---|
| 药物效应动力学(Pharmacodynamics) | 否 | ||
| 体外(“作用机制(MOA)”) | X | X | |
| 体内(“作用机制(MOA)“和"治疗效应(therapeutic effect)”) | X | X | |
| 安全性的药理学(ICH S7A《人用药品安全药理学试验指导原则》和S7B《人用药品延迟心室复极化(QT间期延长)潜在作用的非临床评价指导原则》) | 是 | ||
| 体外(浓度-效应关系) | X | X | |
| 体内(对于CNS,CV,呼吸作用的"剂量反应(dose-response)”) | X | X | |
| “药物代谢动力学(Pharmacokinetics)"(ICH M3(R2)《支持药物进行临床试验和上市的非临床安全性研究指导原则》) | |||
| 体外代谢(跨物种微粒体代谢) | X | NA | 否 |
| 体外血浆蛋白结合 | X | NA | 否 |
| 来自重复给药的GLP毒理学研究的毒代动力学(ICH S3A《毒代动力学指导原则说明:毒性研究中的全身暴露量评价》) | X | X | 是 |
| 遗传毒性标准测试组合(ICH S2(R1)《人用药物遗传毒性试验和结果分析指导原则》) | 是 | ||
| 体外Ames测试 | X | *b | |
| 体外和/或体内哺乳动物细胞染色体损伤评估 | X | *b | |
| 单次给药/剂量范围探索 | 否和是*c | ||
| 啮齿类动物单次给药(可能是MTD研究) | X | NA | |
| 非啮齿类动物单次给药(可能是MTD研究) | X ^e | X ^f | |
| 重复给的药毒理学 ^d(ICH M3(R2)《支持药物进行临床试验和上市的非临床安全性研究指导原则》) | 是 | ||
| 啮齿类动物多次给药 | X | 可选的*f | |
| 非啮齿类动物多次给药 | X ^e | X ^f | |
| 其他研究 | 否 | ||
| 免疫毒性(ICH S8《人用药物免疫毒性研究》) | X | X | |
| 光安全性(ICH S10《药物光安全评价》) | X | X | |
| 滥用倾向 ^h | X | X |
CNS, 中枢神经系统, central nervous system; CV, 心血管, cardiovascular; FIH, 首次人体, first‐in‐human; GLP, 良好的实验室规范, good laboratory practice; ICH, 国际人用药品注册技术协调会, International Conference on Harmonization; MOA, 作用机制, mechanism of action; MTD, 最大耐受剂量, maximum tolerated dose; NA, 不适用, not applicable.
a 请参阅ICH S6(R1)《生物制品的临床前安全性评价》。
b 通常不需要。
c 如果单次给药研究至关重要(即用于支持单次给药"首次人体(FIH)“试验),则应符合GLP标准,这对于大分子而言更为典型。
d 持续时间和给药途径取决于临床试验设计(参考文献6中的表 1)。
e 物种选择取决于新陈代谢与人类的相似性。
f 通常是非人类的灵长类动物或小型猪;取决于靶标的存在和候选药物相对于靶标的相对效力。
g 组织交叉反应性决定了应该研究的物种。如果两种啮齿动物/非啮齿类动物的生物交叉反应,则应研究两种物种。如果该生物制剂仅在一个物种(最常见的是非人类灵长类动物)中具有交叉反应性,则仅对该物种进行研究。如果该生物制品与任何物种都不具有交叉反应性,请考虑使用转基因或替代生物制品。
h 对于基于"作用机制(MOA )"/与已知滥用药物相似的潜在滥用药物。
对于小分子,请参阅"国际人用药品注册技术协调会(ICH)“指南S2(R1)《人用药物遗传毒性试验和结果分析指导原则》中有关遗传毒性测试的建议,该建议不适用于生物制剂。最初,基因突变试验(例如体外 Ames试验)将支持单次给药试验。标准测试组合包括在哺乳动物系统中检测染色体损伤的研究,建议在临床上支持重复给药。
对于安全药理,“国际人用药品注册技术协调会(ICH)“指南S7A《人用药品安全药理学试验指导原则》和S7B《人用药品延迟心室复极化(QT间期延长)潜在作用的非临床评价指导原则》为"国际人用药品注册技术协调会(ICH)” M3(R2)《支持药物进行临床试验和上市的非临床安全性研究指导原则》中描述的核心研究提供了更多详细信息,建议将其用于"首次人体(FIH)“试验,以评估"心血管(CV)",“中枢神经系统,(CNS)“和呼吸系统。最初,进行了体外"心血管(CV)“评估,以确定对QT延长的可能性,这是心律失常的预测因子,方法是评估对” human ether‐a‐go‐go channel(HERG,人类离子通道)”的复合抑制作用。通常也会在体外评估其他在QT延长中起作用的通道,包括钠通道(Nav1.5)和钙通道(Cav 1.2)。此外,为了支持"首次人体(FIH)“试验,在体内建议进行QT评估,通常在剂量范围毒性研究后开始。选择的剂量水平应超过预期的人类暴露水平,但要受所用动物物种的预期耐受性限制。该研究可以独立进行,也可以纳入非啮齿类动物关键毒理学研究。在"国际人用药品注册技术协调会(ICH)” S7A《人用药品安全药理学试验指导原则》的2.9节中,描述了可能不需要进行研究的条件(例如,本地应用的产品)以及对生物制剂的考虑。
体外研究的结果(即遗传毒性和"心血管(CV)“安全性的药理学)将提供起始信息,以基于相对于药效学活性水平的这些安全终点的活性水平确定相对风险。与这些体外评估同时进行的是初步的体内毒性研究,从啮齿类动物开始,以确定动物试验物种的"最大耐受剂量(MTD)“或直至最大可行剂量。非啮齿类动物应选择在药理上最有效的物种(例如,靶序列同源性和相对结合亲和力),对于小分子,应选择与人类代谢相关的物种。通常,犬和非人灵长类动物分别用于小分子和大分子研究。
用于支持"首次人体(FIH)“试验的给药方案和持续时间的关键毒理学研究应符合"良好的实验室规范(GLP)"。剂量范围应较宽,以建立"未见明显毒性反应剂量(NOAEL,no observed adverse effect level)",且其暴露冗余量超过"首次人体(FIH)“试验中预期的最大暴露量。对于小分子,这通常是在啮齿动物和非啮齿类动物中完成的,而对于大分子,仅非啮齿类动物,通常是非人类的灵长类动物就足够了。
最后,应根据药物的光化学特性和该类其他成员的信息进行光毒性的初步评估,这在"国际人用药品注册技术协调会(ICH)” S10《药物光安全评价》中有描述。
在生成上述完整的临床前安全性数据之前,对更早访问人类数据感兴趣的申办方可以考虑进行探索性临床试验,根据"国际人用药品注册技术协调会(ICH)“指南,该试验涉及有限的人体暴露,没有治疗意图,并且不打算进行检查临床耐受性。这些"首次人体(FIH)“研究通常检查"药物代谢动力学(PK)",“药物效应动力学(PD)“和/或生物标志物。指南文件中提供了五种不同的方法以及相应的临床前测试程序,它们的程度有所不同,以支持在治疗范围内进行长达14天的微量给药。
起始剂量选择
在将新药纳入"首次人体(FIH)“试验之前,确定应研究的剂量范围是一个重要目标。通常,剂量是通过利用药理学,毒理学和"药物代谢动力学(PK)“研究获得的所有相关IND启用的临床前结果以及任何具有共同"作用机制(MOA)“的其他疗法可获得的人体经验来确定。最重要的是,起始剂量必须减轻毒性风险,同时平衡引起"药理活性(pharmacologic activity)“的需要,尤其是在给患有更严峻疾病(例如癌症)的患者服用药物时。确定要研究的剂量范围(包括逐步增加的步骤)也很重要,因为当"疗效(efficacy)“成为主要关注因素时,收集到的数据将告知以后在研发中要研究的剂量。
EMA和FDA已发布指南文件,供研究者确定合适的起始剂量时应遵循。EMA指南有助于从临床前开发到早期临床开发,涵盖了"首次人体(FIH)“试验固有的许多风险,并讨论了管理这些风险的缓解策略。FDA指南旨在通过使用公认的安全性基准(从最敏感的毒理学测试物种获得的"未见明显毒性反应剂量(NOAEL)")来确定初始剂量的毒性,以此作为确定合理安全起始剂量的起点。
由于每种新药候选物的独特性以及跨物种转化所必须做出的许多不同假设,因此没有选择正确的"首次人体(FIH)“起始剂量的单一方法。每次将动物的剂量推算到人身上都面临着新的挑战,对于一个候选药物来说,效果很好的方法下一次可能不合适。由于这些原因,很难建立标准的统一方法。但是,监管指南文件确实提供了可遵循的框架。
计算起始剂量的方法是经验性的或机制性的(表 2)。如前所述,最广泛使用的经验方法遵循FDA指南,通过使用异速缩放将"未见明显毒性反应剂量(NOAEL)“转换为人类等效剂量来估算最大安全起始剂量。但是,该方法有其缺点,包括使用某种任意的安全系数来确保起始剂量的安全性,该剂量基于最小的毒性风险而不是药理活性,并且不能解决剂量递增或最大允许剂量。EMA指南概述了一种基于最新模型的更加机制的方法,该模型结合了所有相关的临床前药理学数据,包括人体组织的离体和体外研究。这本指南重点介绍了选择"最低预期生物效应水平(MABEL, minimal anticipated biological effect level)“的方法,因为随着越来越重要的靶向治疗的发展,在预测的药理活性剂量远低于根据"未见明显毒性反应剂量(NOAEL)“认为安全的剂量时,这一点变得越来越重要。
表2. “首次人体(FIH)“临床试验中估计起始剂量的方法
| Method | Advantages | Disadvantages |
|---|---|---|
| MRSD approach (dose‐by‐factor) | Good safety record, easy to calculate | Empirical approach based only on dose, arbitrary safety factor applied, neglects pharmacological activity, and dose escalation |
| Similar MOA | Easy to use; minimal data required | Only applicable to a limited number of drugs, does not account for differences in PK or PD between the two drugs |
| MABEL | Based on pharmacology rather than an empirical scaling factor; safest approach for high‐risk drug candidates with a high degree of species‐specificity or targeting the immune system | Requires more extensive nonclinical data; unclear which nonclinical model/data is most predictive |
| PK model | Accounts for species differences in PK parameters rather than empirical scaling of dose; ability to calculate safety margins; demonstrated to work well for compounds that are eliminated renally and monoclonal antibodies with linear elimination | Neglects species differences in pharmacology (assume concentration‐effect relationship is the same for animals and humans); dependent on accuracy of nonclinical PK and scaling approach |
| PKPD model | One step further than the PK‐guided approach in that it accounts for species differences in both PK and PD; accounts for pharmacologic activity and can support dose escalation | Requires an experienced modeler and extensive nonclinical data |
| 方法 | 优势 | 缺点 |
|---|---|---|
| “推荐的最大安全起始剂量(MRSD)“方法(剂量基于安全系数) | 安全记录良好,易于计算 | 仅基于剂量,所应用的任意安全系数,忽略药理活性和剂量递增的经验性方法 |
| 相似的"作用机制(MOA)” | 使用方便; 所需数据最少 | 仅适用于有限数量的药物,不考虑两种药物之间"药物代谢动力学(PK)“和"药物效应动力学(PD)“的差异 |
| 最低预期生物效应水平(MABEL) | 基于药理学而不是经验比例因子;具有高度物种特异性或针对免疫系统的高风险候选药物的最安全方法 | 需要更广泛的非临床数据; 不清楚哪种非临床模型/数据最能预测 |
| “药物代谢动力学(PK)“模型 | 考虑到"药物代谢动力学(PK)“参数的物种差异,而不是剂量的经验标度;计算安全余量的能力; 被证明对肾脏消除和线性消除的单克隆抗体的化合物有效 | 忽略了药理学上的物种差异(假设动物与人类的浓度-效应关系相同); 取决于非临床PK的准确性和缩放方法 |
| “药物代谢动力学药物效应动力学(PKPD)“模型 | 比"药物代谢动力学(PK)“指导方法更进一步,因为它可以解释"药物代谢动力学(PK)“和"药物效应动力学(PD)“中的物种差异。 解释药理活性并可以支持剂量递增 | 需要经验丰富的建模师和广泛的非临床数据 |
FIH, first‐in‐human, 首次人体; MABEL, minimum anticipated biologic effect level, 最低预期生物效应水平; MOA, mechanism of action, 作用机制; MRSD, maximum recommended safe starting dose, 推荐的最大安全起始剂量; PD, pharmacodynamic, 药物效应动力学; PK, pharmacokinetic, 药物代谢动力学.
“药物代谢动力学/药物效应动力学(PK/PD)“建模在工业中越来越多地使用,它利用浓度-时间过程而不是剂量来从动物推断给人类。“药物代谢动力学/药物效应动力学(PK/PD)“建模提供了一个定量框架,该框架可以通过模拟支持选择预期的治疗剂量范围,从而确定"首次人体(FIH)“临床试验中要研究的剂量上限并支持剂量递增决策。最后,“药物代谢动力学/药物效应动力学(PK/PD)“建模可以解决"药物代谢动力学(PK)“和"药物效应动力学(PD)“的种间差异,从而有可能提高人类预测的准确性。
许多出版物详细介绍了将"药物代谢动力学(PK)“参数从动物缩放到人类的方法。虽然基本"药物代谢动力学(PK)“原则适用于所有的候选药物,但驱动小分子(<1千道尔顿)与大分子吸收,分布,代谢和消除的因素是不同的。因此,物种间缩放和预测人类暴露的方法对于这两种类型的分子通常是不同的。这些方法的全面概述不在本文的讨论范围之内,但是下面将讨论由该分类法分离的基本原理。
小分子
通常,用于种间缩放的方法有两种:i)“基于生理的药代动力学(PBPK)“模型,以及ii)异速缩放。“基于生理的药代动力学(PBPK)“模型提供了缩放小分子"药物代谢动力学(PK)“的更多机制方法。然而,“基于生理的药代动力学(PBPK)“模型需要大量的临床前数据来告知许多模型参数,因此并不经常使用。异速测定法是基于生物学,生理学和解剖学中跨物种相似性而开发的一种不太复杂的经验方法,利用了将生理参数与体型相关联的幂函数。
业已证明,对于小分子,特别是那些主要经历肾脏消除的小分子,异速缩放法非常适用。但是,当小分子药物在"药物代谢动力学(PK)“中表现出较高的跨物种变异性时(例如,由于代谢),这种方法可能效果不佳。其他人则提出了对基于测长法的缩放方法的修改,以提高可预测性。其中包括指数法则法,该法使用最大寿命潜力和脑重量来校正指数,肝血流量,并校正体外代谢清除率。选择的方法将取决于候选药物的"药物代谢动力学(PK)“特性和可用于定标的数据。
“国际人用药品注册技术协调会(ICH)“出版的《抗肿瘤药物非临床评价指导原则》为具有很小治疗窗口(即非常陡峭的浓度安全曲线)的细胞毒药物的经验方法提供了额外的指导。由于癌症患者获得更大的安全风险以实现治疗获益,因此起始剂量的计算是基于对动物产生一定毒性的剂量和方案,只要它不是严重的,不可逆的毒性。该指南建议许多小分子的起始剂量应为适当的起始剂量,该起始剂量应为啮齿动物10%的重度毒性剂量的十分之一,或非啮齿类动物的最高重度毒性剂量的六分之一。重要的是要注意,最高的非严重毒性剂量的定义为,在临床前毒理评估中评估抗癌剂媒介不会产生致命,威胁生命或不可逆毒性的最高剂量水平,它与"未见明显毒性反应剂量(NOAEL)“形成鲜明对比。
大分子
大分子的生物分布通常受极性,电荷和分子大小的限制。大分子通常不是药物代谢酶或药物转运蛋白的底物。相反,大分子的主要消除的途径是肾脏排泄和蛋白降解为氨基酸。幸运的是,这两个过程在整个哺乳动物物种中都是高度保守的。因此,可以应用异速缩放的方法。许多小组已经证明了这一点,这些小组已经证明了使用异速缩放法预测人类的清除率和分布容积的合理准确性。
“单克隆抗体(mAb)“是大分子药物的一个子类,预测单克隆抗体的人类"药物代谢动力学(PK)“的方法主要依赖于非人灵长类动物的PK特征。已经表明,对在猴子中表现出线性"药物代谢动力学(PK)“的"单克隆抗体(mAb)“进行简单的异速缩放可以准确预测人类"药物代谢动力学(PK)“在两倍范围内。然而,对于表现出非线性"药物代谢动力学(PK)“的"单克隆抗体(mAb)",由于靶标介导的药物处置,当低于靶标饱和浓度时,异速伸缩缩放会导致对暴露的高估。如果可以通过实验和文献数据了解相关的靶标介导的药物处置"药物代谢动力学(PK)“参数(例如,靶标结合亲和力常数,基线靶标表达水平和靶标转换速率常数),则表明人类预测是成功的。
上面讨论的指南文件和方法旨在用于全身给药的治疗,尽管类似的原则也应适用于局部给药的治疗(例如局部,吸入和组织内给药)。尽管局部递送增加了目标部位的药物浓度,但涉及将剂量从动物转化为人类的不同假设。这主要是由于以下事实:通常无法评估人的目标部位"药物代谢动力学(PK)“以验证所使用的缩放方法。因此,基于解剖学和生理学的考虑,在非临床模型中评估"药物代谢动力学(PK)“至关重要,该模型被认为最能反映人类,而且通过"基于生理的药代动力学(PBPK)“方法进行种间缩放可能最受益。对每种递送途径的详细讨论不在本文的范围之内。我们推荐读者阅读这些关于眼部局部用药、皮肤局部用药和吸入给药途径的剂量转换的有用文章。
其他设计注意事项
设计"首次人体(FIH)“试验时,在整个研究设计中还需要考虑许多其他因素,包括研究规模,研究人群,剂量递增方案,剂量限制性毒性指标,患者选择和次要目标。此外,通过加快批准程序,例如FDA的快速通道或突破性疗法名称以及EMA的主要地位,候选药物有可能在更短的时间内获得批准。因此,获取关键的临床药理学数据以向处方医生提供有关产品正确使用和风险的信息越来越重要。“首次人体(FIH)“研究为在开发计划中尽早收集此类信息提供了机会。申办方可以纳入对多种剂量,食物效果,相对生物利用度的评估,QT延长和/或"药物相互作用(DDI)“潜力作为"首次人体(FIH)“临床试验的一部分。下文讨论的"首次人体(FIH)“研究的许多要素(如果包含)需要“伞状方案(umbrella protocols)”,以允许进行适应性试验设计,在该试验中,随着可获得的数据,将对研究设计的某些方面进行修改,以最大程度地提高学习效果,而又不损害研究的有效性和质量。研究。图 2说明了具有多个目标的"首次人体(FIH)“研究的可能研究方案。为此,需要制定流程以允许持续不断地及时访问数据,以实现快速审查和决策。
图2
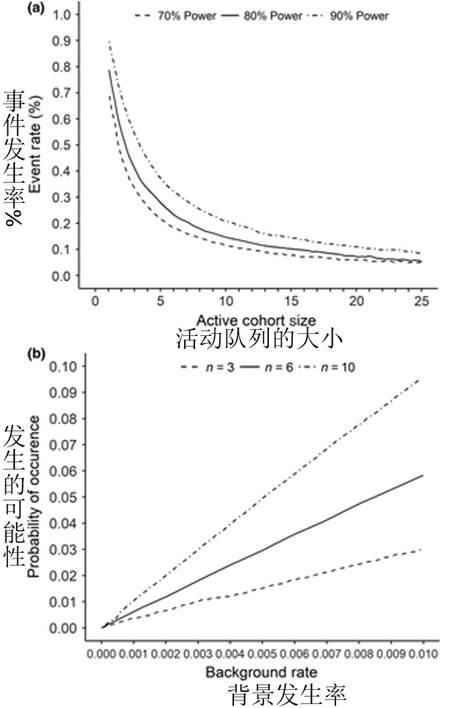
(a)可检测的事件发生率与活性药物队列的样本量和把握度的关系。将队列人数从10人增加到12人几乎无济于事;但从4名受试者增加到6名受试者会导致敏感性显着提高。经Buöen 等人许可转载。(b)在3名,6名和十名受试者的活性药物队列的样本量中,背景发生率与至少一名受试者自发发生事件的概率之间的关系。经Buöen 等人许可转载。 “药物相互作用(DDI)",药品相互作用;MAD,多次给药剂量递增;MTD,最大耐受剂量。
研究规模和纳入安慰剂受试者
大多数"首次人体(FIH)“临床试验都是设盲安慰剂对照研究,每组8至10名受试者以3:1或4:1的比例随机分配,这样六至八名受试者接受了“活性药治疗(active therapy)”,两名受试者接受了"安慰剂(placebo)"。包括多个队列将连续接受更高剂量。但是,这种设计更倾向于根据优先级进行样本量计算,而无需进行后期临床研究中常见的正式假设检验。
“首次人体(FIH)“研究中检测安全信号的能力取决于整个研究中受试者的总数以及每个剂量组的大小,如果给予一个大的群体,实验媒介将导致的实际事件发生率和给定事件的自然背景发生率(非药物相关毒性)。如果假设一个I型错误(不是由药物引起的但将事件归因于某种药物的可能性)和一个II型错误(实际上是由药物引起的给定事件而未检测到事件的可能性),以及由一种疗法引起的一系列事件的发生率,在这种情况下,人们可以画出一条曲线来显示队列规模(N)与观察由于某种疗法在给定的发生率下发生的事件之间的关系。如图2a所示,随着队列规模的增加,可检测的事件发生率呈指数下降,因此在1到6个受试者的范围内增加N会显着提高观察到药物诱发事件的机会。 曲线在N大于10时相对平坦,因此至少观察到由于新疗法引起的事件,队列规模在6至10名受试者之间是合理的。
在"首次人体(FIH)“研究中,只会观察到由于药物引起的非常普遍的事件。对于将在约25%的接受新疗法的受试者中发生的事件,在六个受试者的队列中,有80%的可能性至少一次观察到该事件(图2a)。尽管将队列人数增加到6以上导致观察到给定事件的可能性几乎没有增加,但是样本量越高,观察到不是由于研究药物引起的事件,而仅仅是自发事件发生率的一部分的事件发生的可能性就越大。如图2 b,如果事件的背景发生率为1%,则在6和10的同类群组中观察到该事件的概率分别约为5%和9%。也就是说,在"首次人体(FIH)“研究中,将队列人数增加到6以上不会增加观察药物诱发事件的能力,并导致观察到更多与研究药物无关的事件。
增加总体研究规模的另一个方面是在每个剂量组中纳入安慰剂受试者。纳入安慰剂剂量受试者的理由是不良事件(AE)报告中的感知偏见。尽管样本量很小,但"首次人体(FIH)“方案通常会合并所有安慰剂受试者的研究内分析,以评估活动治疗和安慰剂治疗之间的AE差异。但是,一种思想流派认为,在"首次人体(FIH)“试验中纳入安慰剂受试者的把握度不足,是没有道理或没有用的。据报道,在"首次人体(FIH)“研究中观察到的大多数AE都是“可能相关的”,对因果关系的解释极少,通常情况下,更严重的AE伴随着更大的努力和关注。纳入安慰剂受试者会增加安全性监测委员会要求的程序和文件,以保持对剂量递增决策的设盲影响,从而增加研究的复杂性,这是由于对服用安慰剂的受试者观察到的不良事件,安慰剂的配制和制造以及道德方面的考虑让安慰剂分配的志愿者接受许多潜在的侵入性程序,例如PK抽血。有人可能会说服药前和/或时间匹配的基线测量可代替安慰剂受试者。
研究人群
在健康受试者或患者中进行"首次人体(FIH)“试验的选择应视具体情况而定,要考虑到与参与者安全性有关的所有因素以及所生成数据的价值/质量。这一决定是受研究的候选药物的固有风险以及健康受试者与患者中存在药物靶标的驱动。根据患者的获益与风险,在针对威胁生命的疾病,如果病人所有现有的治疗选择都用光了,那么招募患者而不是健康受试者入组进行较高风险的媒介(即细胞毒性药物)的试验是适当的。另一方面,当合并症或合并用药物难以解释研究结果时,招募健康受试者入组更适合。在某些情况下,在受损的患者中进行测试之前,评估候选新药的安全性和耐受性可能至关重要。但是,值得注意的是,这两个选择不必相互排斥。根据美国国家医学图书馆注册处(clinicaltrials.gov)提供的信息,许多"首次人体(FIH)“研究包括对健康志愿者和患者的评估。如果对健康志愿者进行研究,则入组年龄范围的上限通常限制在45至60之间,以避免在老年人口中常见的许多合并症/伴随药物。
通常在候选药物的"首次人体(FIH)“研究中开始给药时,尚未完全表征其生殖和胚胎/胎儿发育毒性潜能。除非候选药物是为女性健康而设计的,否则由于担心治疗可能对胚胎/胎儿产生不良影响,典型的"首次人体(FIH)“研究会招募男性志愿者,有时还包括没有生育能力的女性。FDA在1993年发布的指南中决定将有潜在生育能力的妇女纳入研究人员,IRB和参加试验的女性中。1998年,美国食品和药物管理局(FDA)提供了有关该主题的最新指南,要求申办方了解PK,安全性和药理反应方面的性别差异;因此,我们希望申办方将女性受试者纳入"首次人体(FIH)“试验中。如果试验中包括有生育能力的妇女,则应仅在持续时间短(例如2周)且对妊娠风险有严格控制的研究中。
剂量和剂量递增方案
为了进一步降低"首次人体(FIH)“试验的风险,在TGN1412事件发生后委托的专家科学小组(又称“达夫”报告)建议按顺序给用药对象,并在各个用药对象之间进行适当的观察。这种做法通常被称为"哨兵给药(sentinel dosing)",并且在单次给药剂量递增(SAD)试验的早期阶段更常采用。对于典型的设盲,安慰剂对照,剂量递增研究,这要求两名受试者以活性药与安慰剂的比例为1:1进行研究,然后再在各自的剂量组中招募其余受试者。根据候选药物的预期AE和MOA,针对每个剂量水平在适当的时间段内执行此操作。最近的EMA指南强调了在每个剂量组中常规使用"哨兵给药(sentinel dosing)"(无论是单次给药还是多次给药),并建议如果不采用这种方法,则应提供“明确的科学依据”。
关于剂量递增的决定要求平衡个体受试者的安全风险与未能定义随后的研究中使用的正确剂量范围的风险,或者更糟却放弃潜在有用药物的开发的风险。为了避免后者,重要的是要了解治疗剂量范围和靶标饱和度,因为在大多数情况下,可以预先确定健康志愿者在"首次人体(FIH)“试验中要研究的预期最大暴露水平。在最近的EMA指南中也对此进行了讨论,在该指南中,健康志愿者的最大暴露量应在估计的人类药效学范围内,因为"最大耐受剂量(MTD)“方法不适用于健康志愿者。2为此,EMA建议实施剂量终止标准,包括研究设计中的最大暴露量(峰值血浆浓度(Cmax)或曲线下面积(AUC)),并且一旦达到个体受试者的阈值,则停止认为符合标准。该新规则将要求申办方仔细定义候选药物的PK/PD关系以及与药效相关的暴露范围。
在患者中进行I期临床试验的重要考虑因素是避免不必要地暴露于亚治疗剂量,同时保持安全性并保持快速升高至治疗剂量范围。剂量发现设计中经常采用的一种策略是在升级到下一个剂量水平之前,以低剂量招募单个受试者以评估安全性和PK。另外,如同抗肿瘤药的情况一样,剂量是使用基于体型大小的剂量而不是固定剂量。基于体型大小的剂量(body size‐based dosing)的实施可能基于以下认识,即受试者间变异性减小,导致反应变异性减小。但是,最近的模拟研究表明,两种给药方法均会导致相似程度的PK和PD变异性,这表明通过体重进行剂量标准化是不必要的步骤。无论采取哪种选项,该策略应提供减少患者间差异,以优化治疗效果以及便利性,依从性和成本效益。
“首次人体(FIH)“肿瘤学试验的主要目标是确定适当的剂量和给药间隔,以在II期试验测试药效效(即推荐的II期剂量)。因此,剂量递增方法支配研究设计,而肿瘤学试验通常属于以下两类之一:基于规则的设计或基于模型的设计。
基于规则的设计基于观察到的目标事件(例如,剂量限制毒性(DLT,dose‐limiting toxicity)),根据预先指定的规则预先指定剂量水平,并逐步增加或降低(即“上下”设计)。最常用的设计是传统的3 + 3设计,该设计以预定的剂量水平处理三名患者的队列(图 3)。剂量递增一直持续到三至六名患者队列中的至少两名患者经历"剂量限制毒性(DLT)"。“最大耐受剂量(MTD)“或推荐的II期剂量是低于有毒水平的剂量。该设计的其他变体已经实现,包括“ 2 + 4”,“ 3 + 3 + 3”和“ 3 + 1 + 1”。另一种基于规则的设计包括药理指导的剂量递增,该剂量递增基于预先指定的与药理活性(例如,显着的肿瘤生长抑制)和/或毒性相关的全身性暴露。通常,一旦达到目标的全身暴露量,该设计就会转换为传统的3 + 3设计。最后,Simon及其同事描述的加速滴定设计42 提供了三种不同的剂量递增设计,直到观察到剂量限制的毒性,此后该设计以40%的剂量步长切换到传统的3 + 3设计。
图3
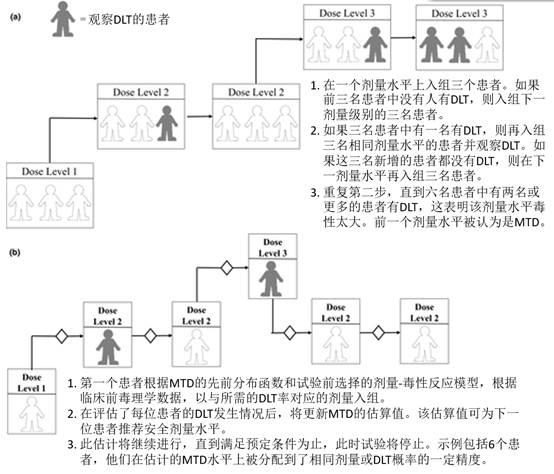
一期肿瘤学研究的研究设计示意图:(a)3 + 3研究设计和(b)持续重新评估模型。
基于规则的设计的一个缺点是它们无法确定剂量。实际上,模拟表明只有35%的患者接受了最佳剂量水平的治疗。相反,55%的患者接受了基于模型设计的最佳剂量的治疗,基于模型的设计,使用统计模型根据预先指定的剂量来分配剂量水平,通过使用所有入组患者的数据来计算"剂量限制毒性(DLT)“的概率,以计算更精确的剂量毒性曲线。基于贝叶斯模型的自适应方法的示例包括连续性重新评估方法(CRM; 图 3),具有剂量过量控制的剂量递增,以及一个CRM修改使用时间-事件终点(time‐to‐event end points)处理晚发性的或CRMs。
这些研究设计是考虑到细胞毒性剂的,其中毒性是主要的决定因素。但是,有许多候选药物具有更大的治疗窗口,因此与"最大耐受剂量(MTD)“相比,需要的思维方式有所不同,但除了毒性之外,还可能需要根据其他终点确定生物学有效剂量。结合毒性和药效的基于贝叶斯的研究设计实例包括EffTox设计或TriCRM。同样重要的是,在阐明创新癌症治疗方法工作队支持的生物学活性剂量范围时,也应考虑PK/PD关系。
还有什么可以考虑的?
传统上,“首次人体(FIH)“研究是SAD研究,与之并行但又滞后的是单独的交错(separate staggered)的多次给药剂量递增(MAD)研究。自从2004年FDA关键路径倡议引入适应性临床试验设计的概念以来,已经开展了更多的SAD / MAD组合研究,与单独的试验相比,估计可将时间表缩短多达12个月。组合设计的吸引力在于,在进行SAD和MAD研究的同时,还可以研究适应性队列中的新剂量,尽管它确实要求预先实施限制性起止标准以确保安全。
“首次人体(FIH)“研究为评估食物和剂型的效应提供了机会,适应性研究设计中经常包含这些信息,以指导进一步的临床开发。当剂量递增达到预期的治疗相关水平时,可以将这些评估纳入研究的SAD组(图 4),这对于PK消除行为非线性的药物尤其重要,无论是基于临床前动物PK数据或从正在进行的试验获得的临床PK数据累积的数据。食物和剂型效应评估均应采用交叉设计进行,以得出有意义的结论。
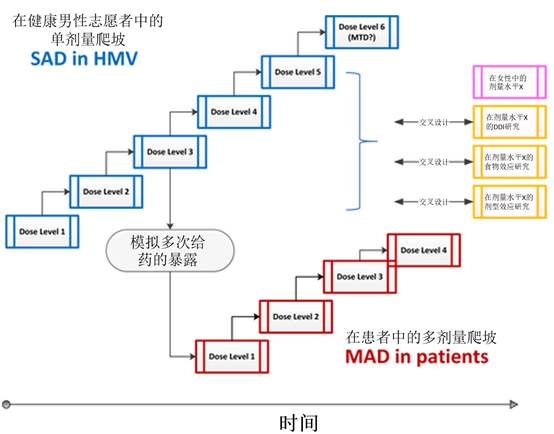
图4
具有多个目标的首次人体试验的示例研究方案。只有在确认健康男性志愿者在相同剂量水平的安全性后,才能启动用于表征治疗相关剂量水平的性别/食物/剂型效应的交叉试验。此阶段的药物相互作用评估通常旨在根据体外数据确定候选药物是有效的细胞色素P450抑制剂的风险。HMV,healthy male volunteers,健康的男性志愿者;MAD,multiple ascending dose,多次给药剂量递增;MTD,maximum tolerated dose,最大耐受剂量;SAD,single ascending dose,单次给药剂量递增。
根据临床前代谢,药物转运和"药物相互作用(DDI)“研究结果,如果候选药物显示出强大的"药物相互作用(DDI)“潜力,尤其是与关键代谢酶(如细胞色素P450 3A4)相关,则谨慎地将"药物相互作用(DDI)“评估纳入"首次人体(FIH)“研究 。 “药物相互作用(DDI)“研究组的设计注意事项应遵循FDA最新指南,以使数据具有临床相关性并提供有益的信息。
FDA指南《治疗性蛋白产品的免疫原性评估》详细介绍了该机构在"首次人体(FIH)“试验中对免疫原性评估的期望,特别是对于高风险蛋白治疗剂。因为动物研究不能预测人类的免疫反应,所以"首次人体(FIH)“研究是申办方评估新生物药物候选药物的免疫原性潜力的第一个机会,这对临床安全性和疗效都有影响。但是,在解释免疫原性数据时应牢记研究人群,因为由于许多因素(例如免疫学状态)可能会改变免疫应答,因此免疫原性谱(mmunogenicity profile)可能或不能代表目标患者群体。
安全监察
“首次人体(FIH)“研究中对安全性的监察给临床医生和申办方带来了悖论。这些试验的典型规模很小,因此非常难以确定安全性信号。除了检测安全信号的统计局限性之外,还很难在临床上确定事件是否是由于新的治疗方式引起的。当研究人群中自发发生许多事件时,将AE归因于实验疗法是有问题的,并且该问题的范围在患者而非健康志愿者中进行的研究中得到了扩大,在健康志愿者中,合并症,疾病进展和其他疗法的持久影响都可以有助于提高信噪比。
“首次人体(FIH)“研究在大多数方面收集的数据与任何临床试验没有什么不同,包括身体检查,生命体征监测,实验室数据(包括血液学,化学和尿液分析)以及在基线和不同时间收集的心电图(ECG)在进行临床试验期间。根据临床前毒理学结果,还可能需要进行专门的检查,例如眼科检查,心理测验,自杀量表的监视或射线照相研究。如果临床前毒理学表明某个候选药物会对心脏传导或对人类离子通道(human ether‐a‐go‐go channels)产生影响,一旦确定了需要进一步开发的剂量或剂量范围,连续的ECG监测和一式三份的ECG结合血浆采样进行PK评估将很有用。实际上,由于"首次人体(FIH)“研究可能是获得用于QTc分析的超剂量用药(supratherapeutic doses)浓度反应数据的唯一机会,因此此类研究带有高质量ECG记录的阴性信号可能消除了进行专门的全面QT延长研究的需要。
当测试新的局部药物候选者时,应使用系统收集的局部反应。当根据临床前信息定义特定的目标AE时,收集比仅描述AE所需的信息更详细的信息将很有用。例如,如果毒理学研究发现在一定暴露量以上会发生异常的肝转氨酶,并且如果在人类受试者中也观察到这种异常,则详细了解近期饮酒和其他药物的病史以及测试可能的混杂原因将有助于确定因果关系,例如针对丙型肝炎病毒,乙型肝炎,人类免疫缺陷病毒,巨细胞病毒和酒精使用指标的血清学筛查。
某些罕见或严重的事件(例如过敏反应,史蒂文斯·约翰逊综合症或急性肝衰竭)几乎总是由活性药物引起的。另外,影响多种信号通路或可能引起免疫或凝血级联反应放大或有释放细胞因子风险的药物在监测和"给药过程(dosing paradigms)“中应格外小心。
许多"首次人体(FIH)“研究的重要成果之一是根据特定严重程度以上的特定AE的发生(称为"剂量限制毒性(DLT)")来确定新治疗实体的"最大耐受剂量(MTD)"。对于其中可能的PK/PD关系知之甚少或一无所知且预期的适应症是具有严重后果的疾病(例如癌症)的治疗领域尤其如此。在此类试验中,目标是确定"最大耐受剂量(MTD)",在尽可能少的剂量水平,并尽量减少可能导致不可接受程度的伤害的剂量。
没有基于"剂量限制毒性(DLT)“发生来确定"最大耐受剂量(MTD)“的标准化设计,但所有设计都包括逐步增加剂量。如上所述,剂量递增可以在受试者内发生或通过队列给药。设计这样的试验的关键的第一步是确定什么毒性是重要的。这些可以是实验室检查(例如肝酶或血清肌酐)或一系列症状(例如严重腹泻)。根据标准化的分级系统(例如美国国家癌症研究所的不良事件通用术语标准)来定义预设的严重性级别。在试验过程中,可能还会出现意料之外的"剂量限制毒性(DLT)"。观察的持续时间也必须定义,在该持续时间期间必须发生的事件以便被认为与实验疗法有关(during which an event has occur in order to be considered related to the experimental therapy)。一般而言,最好进行一项研究以确定将要治疗的患者群体中的"最大耐受剂量(MTD)",另一方面,有时患者会更经常出现某些症状或发现,这是其疾病的一部分,使解释变得困难。在某些情况下,由于已知或怀疑治疗的潜在风险,必须在患者中进行"首次人体(FIH)“研究。此外,必须设置一些潜在"剂量限制毒性(DLT)“的预定义发生率,从而最大程度地减少了归因于治疗的毒性,而实际上却并非如此(译者注,前面的整句话,即上一个句号之后的话)。
这很有用,将PK数据与严重事件或剂量限制事件的进行发生相关。在某些情况下,如果与未经历事件的受试者相比,受试者在极端暴露的情况下AE发展的,则可以在以后的试验中使用此信息来测试基于暴露的剂量是否是一种有利的方法。
“首次人体(FIH)“研究中对不良事件的监测可以通过多种方式来完成,具体取决于感知到的风险。如果预测药物具有低毒性风险,根据 AEs 的发生,有关剂量提升的监测和决策,则可以由申办方(可能与试验的研究者一起)完成。当严重毒性的风险为中度或高度时,应请来对试验结果没有既得利益的专家。通常不需要一个完全独立的数据监察委员会,但是申办方和相关治疗领域或临床学科的外部专家之间的共同努力通常有助于确定是否已达到潜在的不安全剂量。但是,在某些情况下,如果出于某种原因以安慰剂对照双盲方式进行"首次人体(FIH)“研究,并且高度关注新药可能毒性过大,则可能需要一个独立的数据监察委员会。这样的委员会可以了解不设盲的数据,以便更容易识别药物引起的作用。在所有情况下,在启动 “首次人体(FIH)” 研究之前设计安全检查计划非常重要。该计划的目的是阐明将怎样收集数据以及收集那些数据,将怎样审查以及在何时审查,以及可能的建议。例如,在毒理学研究表明可能发生特定类型的医学上严重的毒性的情况下,应制定研究中个体受试者将需要停止给药或完全停止研究的规则规定。
生物标志物评估
“首次人体(FIH)“研究意味着在人类上获得生物学读数的第一个机会。生物标志物与药物治疗相结合的测量在药物开发和药物上市申请批准后的临床实施中都可能很重要。生物标志物的读数可以帮助指导药物开发:从发现,到通过靶点参与度评估,到诊断,再到在日常临床实践中对精准医学和伴随诊断的承诺。沿着这条道路,在"首次人体(FIH)“研究中实施生物标志物计划是迈向生物标志物效用的关键一步,因为它可以确认来自临床前或观察性临床研究的假设。存在许多在早期临床研究中进行测试的生物标记物的例子,这些生物标记物已继续用于临床诊断。这在肿瘤学领域尤为明显,因为预测性生物标志物通常用于患者筛查和风险评估,例如BRCA1 / 2和前列腺特异性抗原,以及预后和治疗决策,例如用于曲妥珠单抗治疗的人表皮生长因子受体2。另外,使用"首次人体(FIH)“研究中优先选择的候选生物标志物也可以支持MOA假设,从而导致进一步的概念验证(POC)研究或特定疾病的新靶标。
通常认为药理或疾病状态的生物标志物是可在血清或血浆中轻易测量的可溶性全身性因子。然而,有许多终点或患者分层因素也可以测量,例如遗传,可溶性细胞表面或组织学标记物。临床药理学家/科学家应了解文献,以及体外和临床前药理模型,以确定是否应在"首次人体(FIH)“研究中包括合适的候选生物标志物。在临床前动物或体外模型以及安全性评估期间,应通过调节候选生物标志物来证明对药物药理学有扎实的理解。评估生物标志物的决定应包括对各种成功因素的评估,例如全面的文献综述,生物分析方法开发,可行性和验证,临床采样策略,数据减少和分析策略(data reduction and analysis strategy),以及如何以用有意义的方式将生物标志物数据纳入临床数据库的计划。还应进行监管评估。“首次人体(FIH)“的数据将仅用于支持进入II期POC?另一方面,是否将使用生物标志物数据为进一步的生物标志物研究奠定基础,通过后期临床研究以及临床诊断测试,例如许可的筛查工具或伴随诊断程序?这些问题的答案将有助于确定适合目的的监管需求的生物标志物方法和数据处理。在规划生物标记物实施方案时,必须进行整体思考,因为一个"首次人体(FIH)“生物标记物数据集不仅可能对单个药物开发项目有价值,而且对将要开发多种候选药物的疾病适应症有用。
一旦确定需要在"首次人体(FIH)“研究中评估生物标志物,就必须证明在临床试验中进行生物标志物分析的可行性。根据所选的生物标志物以及有关采样,测量和分析数据所需方法的可用信息,这可能是一个简单的基于纸张的练习,或者可能涉及大量的实验室工作,以证明所提出的方法可以适当地测量生物标志物。在后一种情况下,评估生物标记的决定将被最佳地提前足够长的时间,以使得能够开发出可靠的生物分析方法,并且在计划"首次人体(FIH)“时应考虑到这一点。如果对生物标记物的稳定性有任何了解,例如血清中给定的蛋白质,那么就可以在"首次人体(FIH)“研究结束时计划进行批次分析,并与"首次人体(FIH)“研究同时进行有风险的方法验证。与开发定制的配体结合方法相比,其他生化测量方法(例如液相色谱-质谱法或脂质或其他代谢物的自动化临床分析仪)可能更容易获得。不管最终的生物分析方法如何,在获得比血清或血浆难及的组织或样品时可能会有挑战。具有挑战性的基质(例如粪便样本,眼液,粘膜分泌物,组织活检组织和外科手术副产物)可以并且应考虑用于生物标志物分析。
生物标记物位于法规遵从性区域中,使临床开发团队可以决定所需水平的方法特征和记录。至少,应通过测试在目标基质或相关替代基质中独立制备的质量控制样品来进行证明精密度和准确性的鉴定。在最佳情况下,“首次人体(FIH)“生物标志物生物分析测定应是一种经过充分验证的方法,包括先验确定该方法的可量化范围和基质干扰,然后对测定的精度,准确性,选择性,稳健性,内源性进行基于计划的评估健康状态和疾病状态基质的水平,平行性以及在模拟样品处理的存储条件下的分析物稳定性。通过权衡临床开发计划的目标以及生物标记物如何为该计划提供最大价值,可以最好地评估针对给定生物标记物分析的适用合规水平。例如,如果将生物标志物用于评估II期POC临床反应以支持III期决策,则应实施经过充分验证的方法。但是,如果"首次人体(FIH)“中的生物标志物旨在支持药物输送技术的计划通过/不通过决定,则可能只需要进行合格的测定。无论目的如何,该方法的调节和生物分析特性均应符合程序的需求。
与评估分析开发需求以适应给定"首次人体(FIH)“生物标志物测量的目的类似,应前瞻性地定义生物标志物数据分析计划。例如,基因表达研究通常依靠管家基因(housekeeping genes,为维持细胞的基本生命活动所需而时刻都在表达的基因)来标准化给定样品和基质(例如眼泪或尿液)中的细胞数量,其浓度或体积因生物学差异而有所不同,因此可能需要内部归一化因子,例如总蛋白或肌酐。监管要求还可以定义应如何处理数据。如果"首次人体(FIH)“生物标志物评估本质上是探索性的,则简单的电子表格分析可能就足够了。但是,如果要为生物标记物制定规范的开发路径,则需要符合临床数据交换标准协会标准的数据标准。
全球考虑
影响"首次人体(FIH)“试验研究地点/国家/地区选择的因素很多,包括申办方的经营地点,监管要求,入组难易程度和目标患者人群的分布。一旦决定,与当地卫生当局的早期接触和在战略上适时的互动对于"首次人体(FIH)“研究的成功至关重要。但是,在进行此类参与之前,如果有指南文件,则对监管机构的期望有透彻的了解将更好地帮助申办方,并确保进行富有成效的讨论。在没有明确指南的情况下,可能会聘请经验丰富的当地临床合同研究组织,临床开发顾问和主要意见领袖来联络和支持研究。
过去,“国际人用药品注册技术协调会(ICH)“国家的监管机构是最早评估新化学实体和新生物产品的机构之一(世界卫生组织(WHO),“ WHO会议报告:“国际人用药品注册技术协调会(ICH)“准则在非’国际人用药品注册技术协调会(ICH)“国家中的实施影响’ ”)。近年来,包括在非"国际人用药品注册技术协调会(ICH)“国家(如澳大利亚)进行的"首次人体(FIH)“研究在内的早期临床试验的数量已大大增加。尽管非"国际人用药品注册技术协调会(ICH)“国家通常采用"国际人用药品注册技术协调会(ICH)"‐Good Clinical Practice(设定全球临床试验运行标准的"国际人用药品注册技术协调会(ICH)"),人们认为不严格的要求和更快开始"首次人体(FIH)“试验的批准程序可能是增加费用的因素。重要的是要注意,即使所生成的临床数据可以被"国际人用药品注册技术协调会(ICH)“监管机构接受,但只要确保研究质量,药物质量或临床前安全性方面的数据差距仍可能会阻止申办方在"国际人用药品注册技术协调会(ICH)“国家继续进行临床开发。
结论
总之,“首次人体(FIH)“研究要求跨职能的合作才能成功。适应性研究设计允许回答许多问题,这将有利于下游临床开发。“首次人体(FIH)“研究中的安全数据监察和收集与后期研究相似,需要制定明确的计划来监察和评估安全数据。
资金
这项工作没有收到任何资金。
利益冲突
作者宣称对这项工作没有任何利益冲突。
参考文献:
1 食品药品监督管理局药品评估与研究中心。 估计成人健康志愿者治疗的初始临床试验的最大安全起始剂量。< https://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf >引用(2005)。2008年3月2日访问
2 欧洲药品管理局。 鉴定和减轻使用研究用药物进行首次人类和早期临床试验风险的策略指南。
<http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_001001.jsp&mid=WC0b01ac0580029570 >(2018)。于2018年3月2日访问。
3 A. S. Faqi编辑。 临床前药物开发的毒理学综合指南。(学术出版社: 伦敦,2013年)。 885页。
4 Gad,SC。 药品和医疗器械的安全性评价。(美国Springer: 马萨诸塞州波士顿,2011年)。
5 Butler,L. D 等。 支持安全临床试验的当前非临床试验范例:IQ Consortium DruSafe的观点。Regul。毒药。Pharmacol。 87, S1 - S15(2017)。
6 国际人用药品注册技术协调会。 关于进行人类临床试验和药品M3(R2)的销售授权的非临床安全性研究指南。
<https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M3_R2/Step4/M3_R2__Guideline.pdf >(2009)访问2018年4月12日。
7 生物技术衍生药品S6(R1)的临床前安全性评估。
<http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S6_R1/Step4/S6_R1_Guideline.pdf >(2011)。于2018年6月19日访问。
8 抗癌药物S9的非临床评估。
<http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S9/Step4/S9_Step4_Guideline.pdf >(2009年)。于2018年6月29日访问。
9 供人类使用的药物的遗传毒性测试和数据解释指南S2(R1)。
<http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S2_R1/Step4/S2R1_Step4.pdf >(2011)。于2018年4月12日访问。
10 药品的光安全性评估S10。
<http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S10/S10_Step_4.pdf >(2013)。于2018年4月12日访问。
11 Agoram,BM 在高危生物制剂的首次人体试验中,使用药代动力学/药效学模型来开始剂量选择。Br。J.临床 Pharmacol。 67(2), 153 – 160(2009)。
12 Reigner,BG和Blesch,KS 估计进入人体的起始剂量:原理和实践。欧元。J.临床 Pharmacol。 57(12), 835 – 845(2002)。
13 Bender,BC,Schindler,E.&Friberg,LE 肿瘤学中的人群药代动力学-药效学建模:一种预测临床反应的工具:临床肿瘤学中的人群药代动力学-药效学建模。Br。J.临床 Pharmacol。 79(1), 56 – 71(2015)。
14 Huh,Y.,Smith,DE和Rose Feng,M. 种间定标和人类清除率的预测:小分子和大分子药物的比较。Xenobiotica 41(11), 972 – 987(2011)。
15 Mahmood,I.&Balian,JD 种间缩放:预测人类中药物的清除率。三种不同的方法。Xenobiotica 26(9), 887 – 895(1996)。
16 Dong,JQ 等。 单克隆抗体对人体药代动力学的定量预测:猴作为单一物种的回顾性分析,用于首次人类预测。临床 药代动力学。 50(2), 131 – 142(2011)。
17 Singh,AP 等。 展示目标介导特性的mAb的人体药代动力学定量预测。AAPS J. 17(2), 389 - 399(2015)。
18 Bischoff,KB 将药代动力学应用于癌症化学疗法的一些基本考虑。癌症化学疗法。代表。 59(4), 777 - 793(1975)。
19 Boxenbaum,H. 种间缩放,异速测量,生理时间和药代动力学基本计划。J.药代动力学。生物制药。 10(2), 201 - 227(1982)。
20 Nagilla,R.和Ward,KW 对校正因子在从大鼠,狗和猴子到人的清除率的异速预测中的作用进行了全面分析。J.药物 科学 93(10), 2522 – 2534(2004)。
21 Lave,T。 等人。 波生坦(一种新的内皮素受体拮抗剂)的种间缩放和将体外数据整合到异速缩放中。药 Res。 13(1), 97 – 101(1996)。
22 Tabrizi,M.,Bornstein,GG和Suria,H 。治疗性单克隆抗体在健康和疾病中的生物分布机制。AAPS J. 12(1), 33 - 43(2010)。
23 Mordenti,J.,Chen,SA,Moore,JA,Ferraiolo,BL&Green,JD 种间比例缩放五个治疗性蛋白质的清除率和分布数据量。药 Res。 8(11), 1351 – 1359(1991)。
24 Mahmood,I. 抗体的药代动力学异形缩放:在首次人类剂量估计中的应用。J.药物 科学 98(10), 3850 – 3861(2009)。
25 Wang,W.&Prueksaritanont,T. 对人类治疗性蛋白质清除率的预测:重新探讨了简单的异度缩放法。生物制药。药物处置。 31(4), 253 - 263(2010)。
26 Luu,KT,Bergqvist,S.,Chen,E.,Hu-Lowe,D.和Kraynov,E. 一种基于模型的方法来预测具有靶标介导药物配置的单克隆抗体的人药代动力学。J.Pharmacol。经验值 那个 341(3), 702 – 708(2012)。
27 Durairaj,C.,Shen,J.和Cherukury,M .基于机制的平移药代动力学-药效学模型可预测青光眼或高眼压患者的眼压降低效果。药 Res。 31(8), 2095 – 2106(2014)。
28 Lehman,PA,Raney,SG和Franz,TJ人的 经皮吸收:体内外相关性。皮肤药理学。生理。 24(4), 224 – 230(2011)。
29 Phillips,JE 吸入从啮齿动物到人的有效剂量转换:呼吸系统疾病临床标准的回顾性分析。Pharmacol。那个 178, 141 - 147(2017)。
30 Buöen,C.,Holm的,S.&汤姆森,MS 在基于实验室数据相予剂量递增临床试验的队列大小的评价。J.临床 Pharmacol。 43(5), 470 – 476(2003)。
31 Iasonos,A。 等。 在第一阶段试验中,非药物毒性对估计最大耐受剂量的影响。临床 癌症研究。 18(19), 5179 - 5187(2012)。
32 Sackett,DL&Gent,M . 关于临床试验中计数和归因事件的争议。N. Engl。J. Med。 301(26), 1410 – 1412(1979)。
33 Parasrampuria,DA&Benet,LZ 尚无理由将安慰剂和盲法用于递增剂量的首次人体研究和其他动力不足的1期研究,总的来说是没有用的。基本临床。Pharmacol。毒药。 117(1), 44 – 51(2015)。
34 Liu,KA,DiPietro Mager,NA。 妇女参与临床试验的历史和未来意义。药 实践 14(1), 708(2016)。
第35 天, 达夫(M. Duff)的报告呼吁改变药物测试的方式。BMJ 333(7581), 1240 – 1242(2006)。
36 Bai,S。 等。 指导合理剂量的单克隆抗体。临床 药代动力学。 51(2), 119 – 135(2012)。
37 Wang DD,Zhang,S.,Zhao,H.,Men,AY&Parivar,K . 在成人临床试验中单克隆抗体的固定剂量与基于体型的剂量。J.临床 Pharmacol。 49(9), 1012 – 1024(2009)。
38 Hempel,G.&Boos,J. 基于固定剂量的药物与基于身体表面积的抗癌药物的剂量:存在差异。肿瘤学家 12(8), 924 – 926(2007)。
39 Ananthakrishnan,R.和Menon,S .肿瘤学临床试验设计:综述。暴击 牧师 血红素 88(1), 144 – 153(2013)。
40 Rogatko,A.,Schoeneck,D.,乔纳斯,W.,Tighiouart,M.,Khuri,FR&波特A. 创新设计进入I期试验翻译。J.临床 Oncol。 25(31), 4982 - 4986(2007)。
41 Collins,JM,Grieshaber,CK和Chabner,BA 基于临床前药物开发的药理学指导的I期临床试验。J.纳特尔 癌症研究所 82(16), 1321 - 1326(1990)。
42 Simon,R.,Freidlin,B.,Rubinstein,L.,Arbuck,SG,Collins,J.和Christian,MC 肿瘤一期临床试验的加速滴定设计。J.纳特尔 癌症研究所 89(15), 1138 – 1147(1997)。
43 Ivy,SP,Siu,LL,Garrett-Mayer,E.&Rubinstein,L. 集中于安全性,效率和特定患者人群的1期临床试验设计方法:美国国家临床试验设计工作组的报告癌症研究所研究药物指导委员会。临床 癌症研究。 16(6), 1726 – 1736(2010)。
44 Babb,J.,Rogatko,A.和Zacks,S. 癌症I期临床试验:有效的剂量递增和过量控制。统计 中 17(10), 1103 - 1120(1998)。
45 Normolle,D.&Lawrence,T. 使用事件持续时间重新评估方法设计具有晚期发作毒性的剂量递增试验。J.临床 Oncol。 24(27), 4426 - 4433(2006年)。
46 Thall,PF&Cook,JD 基于功效-毒性权衡的剂量寻找。Biometrics 60(3), 684 – 693(2004)。
47 Zhang,W.,Sargent,DJ&Mandrekar,S. 一种兼具毒性和功效的适应性剂量寻找设计。统计 中 25(14), 2365 – 2383(2006)。
48 Booth,CM,Calvert,AH,Giaccone,G.,Lobbezoo,MW,Seymour,LK&Eisenhauer,EA Endpoints和针对靶向抗癌治疗的第一阶段研究中的其他注意事项:创新方法开发工作组的建议癌症疗法(MDICT)。欧元。癌症杂志 44(1), 19 - 24(2008)。
49 Francis,M.,Basque,J.,Legault,E.,Rufiange,M.,Sicard,E.&Lefebvre,M .涉及先后顺序单次和多次递增剂量队列研究的综合人类首次研究设计。一期临床试验的趋势。临床 Pharmacol。那个 93, S20 – S21(2013)。
50 食品药品监督管理局药品评估研究中心。 体外代谢和转运蛋白介导的药物相互作用研究(指导草案)。
<https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM581965.pdf >(2017)访问于2018年3月26日。
51 食品药品监督管理局药品评估与研究中心。 临床药物相互作用研究-研究设计,数据分析和临床意义(指导草案)。
<https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf >(2017年)。于2018年3月26日访问。
52 治疗性蛋白质产品的免疫原性评估。
<https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM338856.pdf >(2014)。于2018年6月29日访问。
53 E14非抗心律不齐药物的qt / qtc间隔延长和心律失常潜力的临床评估-问答(R3)行业指南。
<https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073161.pdf >(2017)。于2018年4月12日访问。
54 Cutler,NR 等。 定义最大耐受剂量:研究人员,学术界,行业和监管部门的观点。J.临床 Pharmacol。 37(9), 767 – 783(1997)。
55 Cutler,NR和Sramek,JJ 研究人员对MTD的看法:MTD定义的实际应用–是否加快了发展? J.临床 Pharmacol。 40(11), 1184 - 1187年(2000)。讨论1202-1204。
56 食品药品监督管理局药品评估与研究中心。 临床试验发起人建立和运行临床试验数据监测委员会的指南。
<https://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm127073.pdf >(2006)。于2018年4月12日访问。
57 Goossens,N.,Nakagawa,S.,Sun,X.&Hoshida,Y. 癌症生物标志物的发现和验证。翻译 癌症研究。 4(3), 256 – 269(2015)。
58 阿诺德,我,布斯,B。,金,L。和雷,C。 研讨会报告:水晶之城六—生物标记物的生物分析方法验证。AAPS J. 18(6), 1366 - 1372(2016)。
59 世界卫生组织。 在非"国际人用药品注册技术协调会(ICH)“国家中实施"国际人用药品注册技术协调会(ICH)“指南的影响。
<http://apps.who.int/medicinedocs/pdf/h2993e/h2993e.pdf >(2001年)。于2018年4月12日访问。
60 Frost&Sullivan。 澳大利亚:早期临床试验的首选目的地。
<https://ww2.frost.com/files/6514/7374/3781/Novotech_WP_20160701_v2.1.pdf >(2016)。于2018年4月12日访问。
61 Lang,TA 等。 在资源有限的环境中进行临床研究:增强研究能力并共同努力,以简化试验。(编者 CH King)。PLoS Negl。放下 Dis。。 4(6), e619(2010)。
62 国际人用药品注册技术协调会。 人类药物S7A的安全药理研究。
<http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S7A/Step4/S7A_Guideline.pdf >(2000)。于2018年4月12日访问。
63 国际人用药品注册技术协调会 延迟心室复极化(QT间隔延长)S7B的潜力的非临床评估。
<http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S7B/Step4/S7B_Guideline.pdf >(2005年)。于2018年4月12日访问。
64 国际人用药品注册技术协调会关于 毒物动力学的指导说明:毒性研究S3A中的全身暴露评估。
<http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S3A/Step4/S3A_Guideline.pdf >(1994年)。于2018年4月12日访问。
65 国际 人类药物协调免疫毒性研究会议S8。
<http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S8/Step4/S8_Guideline.pdf >(2005年)。于2018年4月12日访问。
英语版参考文献列表:
1 Food and Drug Administration, Center for Drug Evaluation and Research. Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. https://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf Cited (2005). Accessed March 2, 2008
Google Scholar
2 European Medicines Agency. Guideline on strategies to identify and mitigate risks for first‐in‐human and early clinical trials with investigational medicinal products. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_001001.jsp&mid=WC0b01ac0580029570 (2018). Accessed March 2, 2018.
Google Scholar
3 A. S. Faqi, ed. A Comprehensive Guide to Toxicology in Preclinical Drug Development. ( Academic Press: London, 2013). 885 p.
Google Scholar
4Gad, S. C. Safety Evaluation of Pharmaceuticals and Medical Devices. ( Springer US: Boston, MA, 2011).
Google Scholar
5Butler, L. D et al. Current nonclinical testing paradigms in support of safe clinical trials: an IQ Consortium DruSafe perspective. Regul. Toxicol. Pharmacol. 87, S1– S15 (2017).
CrossrefPubMedWeb of Science®Google Scholar
6 International Conference on Harmonisation. Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals M3(R2). https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M3_R2/Step4/M3_R2__Guideline.pdf (2009) Accessed April 12, 2018.
Google Scholar
7 Preclinical safety evaluation of biotechnology‐derived pharmaceuticals S6(R1). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S6_R1/Step4/S6_R1_Guideline.pdf (2011). Accessed June 19, 2018.
Google Scholar
8 Nonclinical evaluation for anticancer pharmaceuticals S9. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S9/Step4/S9_Step4_Guideline.pdf (2009). Accessed June 29, 2018.
Google Scholar
9 Guidance on genotoxicity testing and data interpretation for pharmaceuticals intended for human use S2(R1). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S2_R1/Step4/S2R1_Step4.pdf (2011). Accessed April 12, 2018.
Google Scholar
10 Photosafety evaluation of pharmaceuticals S10. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S10/S10_Step_4.pdf (2013). Accessed April 12, 2018.
Google Scholar
11Agoram, B.M. Use of pharmacokinetic/ pharmacodynamic modelling for starting dose selection in first‐in‐human trials of high‐risk biologics. Br. J. Clin. Pharmacol. 67( 2), 153– 160 (2009).
Wiley Online LibraryCASPubMedWeb of Science®Google Scholar
12Reigner, B.G. & Blesch, K.S. Estimating the starting dose for entry into humans: principles and practice. Eur. J. Clin. Pharmacol. 57( 12), 835– 845 (2002).
CrossrefCASPubMedWeb of Science®Google Scholar
13Bender, B.C., Schindler, E. & Friberg, L.E. Population pharmacokinetic‐pharmacodynamic modelling in oncology: a tool for predicting clinical response: population pharmacokinetic‐pharmacodynamic modelling in clinical oncology. Br. J. Clin. Pharmacol. 79( 1), 56– 71 (2015).
Wiley Online LibraryCASPubMedWeb of Science®Google Scholar
14Huh, Y., Smith, D.E. & Rose Feng, M. Interspecies scaling and prediction of human clearance: comparison of small‐ and macro‐molecule drugs. Xenobiotica 41( 11), 972– 987 (2011).
CrossrefCASPubMedWeb of Science®Google Scholar
15Mahmood, I. & Balian, J.D. Interspecies scaling: predicting clearance of drugs in humans. Three different approaches. Xenobiotica 26( 9), 887– 895 (1996).
CrossrefCASPubMedWeb of Science®Google Scholar
16Dong, J.Q. et al. Quantitative prediction of human pharmacokinetics for monoclonal antibodies: retrospective analysis of monkey as a single species for first‐in‐human prediction. Clin. Pharmacokinet. 50( 2), 131– 142 (2011).
CrossrefCASPubMedWeb of Science®Google Scholar
17Singh, A.P. et al. Quantitative prediction of human pharmacokinetics for mAbs exhibiting target‐mediated disposition. AAPS J. 17( 2), 389– 399 (2015).
CrossrefCASWeb of Science®Google Scholar
18Bischoff, K.B. Some fundamental considerations of the applications of pharmacokinetics to cancer chemotherapy. Cancer Chemother. Rep. 59( 4), 777– 793 (1975).
CASPubMedGoogle Scholar
19Boxenbaum, H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J. Pharmacokinet. Biopharm. 10( 2), 201– 227 (1982).
CrossrefCASPubMedWeb of Science®Google Scholar
20Nagilla, R. & Ward, K.W. A comprehensive analysis of the role of correction factors in the allometric predictivity of clearance from rat, dog, and monkey to humans. J. Pharm. Sci. 93( 10), 2522– 2534 (2004).
Wiley Online LibraryCASPubMedWeb of Science®Google Scholar
21Lave, T. et al. Interspecies scaling of bosentan, a new endothelin receptor antagonist and integration of in vitro data into allometric scaling. Pharm. Res. 13( 1), 97– 101 (1996).
CrossrefCASPubMedWeb of Science®Google Scholar
22Tabrizi, M., Bornstein, G.G. & Suria, H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J. 12( 1), 33– 43 (2010).
CrossrefCASPubMedWeb of Science®Google Scholar
23Mordenti, J., Chen, S.A., Moore, J.A., Ferraiolo, B.L. & Green, J.D. Interspecies scaling of clearance and volume of distribution data for five therapeutic proteins. Pharm. Res. 8( 11), 1351– 1359 (1991).
CrossrefCASPubMedWeb of Science®Google Scholar
24Mahmood, I. Pharmacokinetic allometric scaling of antibodies: application to the first‐in‐human dose estimation. J. Pharm. Sci. 98( 10), 3850– 3861 (2009).
Wiley Online LibraryCASPubMedWeb of Science®Google Scholar
25Wang, W. & Prueksaritanont, T. Prediction of human clearance of therapeutic proteins: simple allometric scaling method revisited. Biopharm. Drug Dispos. 31(4), 253‐ 263 (2010).
Wiley Online LibraryCASPubMedWeb of Science®Google Scholar
26Luu, K.T., Bergqvist, S., Chen, E., Hu‐Lowe, D. & Kraynov, E. A model‐based approach to predicting the human pharmacokinetics of a monoclonal antibody exhibiting target‐mediated drug disposition. J. Pharmacol. Exp. Ther. 341( 3), 702– 708 (2012).
CrossrefCASPubMedWeb of Science®Google Scholar
27Durairaj, C., Shen, J. & Cherukury, M. Mechanism ‐ based translational pharmacokinetic ‐ pharmacodynamic model to predict intraocular pressure lowering effect of drugs in patients with glaucoma or ocular hypertension. Pharm. Res. 31( 8), 2095– 2106 (2014).
CrossrefCASPubMedWeb of Science®Google Scholar
28Lehman, P.A., Raney, S.G. & Franz, T.J. Percutaneous absorption in man: in vitro‐in vivo correlation. Skin Pharmacol. Physiol. 24( 4), 224– 230 (2011).
CrossrefCASPubMedWeb of Science®Google Scholar
29Phillips, J.E. Inhaled efficacious dose translation from rodent to human: a retrospective analysis of clinical standards for respiratory diseases. Pharmacol. Ther. 178, 141– 147 (2017).
CrossrefCASPubMedWeb of Science®Google Scholar
30Buöen, C., Holm, S. & Thomsen, M.S. Evaluation of the cohort size in phase I dose escalation trials based on laboratory data. J. Clin. Pharmacol. 43( 5), 470– 476 (2003).
Wiley Online LibraryCASPubMedWeb of Science®Google Scholar
31Iasonos, A. et al. The impact of non‐drug‐related toxicities on the estimation of the maximum tolerated dose in phase I trials. Clin. Cancer Res. 18( 19), 5179– 5187 (2012).
CrossrefCASPubMedWeb of Science®Google Scholar
32Sackett, D.L. & Gent, M. Controversy in counting and attributing events in clinical trials. N. Engl. J. Med. 301( 26), 1410– 1412 (1979).
CrossrefPubMedWeb of Science®Google Scholar
33Parasrampuria, D.A. & Benet, L.Z. Inclusion of placebos and blinding for ascending dose first‐in‐human studies and other underpowered phase 1 studies has not been justified and on balance is not useful. Basic Clin. Pharmacol. Toxicol. 117( 1), 44– 51 (2015).
Wiley Online LibraryCASPubMedWeb of Science®Google Scholar
34Liu, K.A., DiPietro Mager, NA. Women’s involvement in clinical trials: historical perspective and future implications. Pharm. Pract. 14( 1), 708 (2016).
CrossrefPubMedGoogle Scholar
35Day, M. Duff’s report calls for changes in way drugs are tested. BMJ 333( 7581), 1240– 1242 (2006).
CrossrefGoogle Scholar
36Bai, S. et al. A guide to rational dosing of monoclonal antibodies. Clin. Pharmacokinet. 51( 2), 119– 135 (2012).
CrossrefCASPubMedWeb of Science®Google Scholar
37Wang, D.D., Zhang, S., Zhao, H., Men, A.Y. & Parivar, K. Fixed dosing versus body size‐based dosing of monoclonal antibodies in adult clinical trials. J. Clin. Pharmacol. 49( 9), 1012– 1024 (2009).
Wiley Online LibraryCASPubMedWeb of Science®Google Scholar
38Hempel, G. & Boos, J. Flat‐fixed dosing versus body surface area based dosing of anticancer drugs: there is a difference. Oncologist 12( 8), 924– 926 (2007).
Wiley Online LibraryCASPubMedWeb of Science®Google Scholar
39Ananthakrishnan, R. & Menon, S. Design of oncology clinical trials: a review. Crit. Rev. Oncol. Hematol. 88( 1), 144– 153 (2013).
CrossrefPubMedWeb of Science®Google Scholar
40Rogatko, A., Schoeneck, D., Jonas, W., Tighiouart, M., Khuri, F.R. & Porter, A. Translation of innovative designs into phase I trials. J. Clin. Oncol. 25( 31), 4982– 4986 (2007).
CrossrefPubMedWeb of Science®Google Scholar
41Collins, J.M., Grieshaber, C.K. & Chabner, B.A. Pharmacologically guided phase I clinical trials based upon preclinical drug development. J. Natl. Cancer Inst. 82( 16), 1321– 1326 (1990).
CrossrefCASPubMedWeb of Science®Google Scholar
42Simon, R., Freidlin, B., Rubinstein, L., Arbuck, S.G., Collins, J. & Christian, M.C. Accelerated titration designs for phase I clinical trials in oncology. J. Natl. Cancer Inst. 89( 15), 1138– 1147 (1997).
CrossrefCASPubMedWeb of Science®Google Scholar
43Ivy, S.P., Siu, L.L., Garrett‐Mayer, E. & Rubinstein, L. Approaches to phase 1 clinical trial design focused on safety, efficiency, and selected patient populations: a report from the Clinical Trial Design Task Force of the National Cancer Institute Investigational Drug Steering Committee. Clin. Cancer Res. 16( 6), 1726– 1736 (2010).
CrossrefPubMedWeb of Science®Google Scholar
44Babb, J., Rogatko, A. & Zacks, S. Cancer phase I clinical trials: efficient dose escalation with overdose control. Stat. Med. 17( 10), 1103– 1120 (1998).
Wiley Online LibraryCASPubMedWeb of Science®Google Scholar
45Normolle, D. & Lawrence, T. Designing dose‐escalation trials with late‐onset toxicities using the time‐to‐event continual reassessment method. J. Clin. Oncol. 24( 27), 4426– 4433 (2006).
CrossrefCASPubMedWeb of Science®Google Scholar
46Thall, P.F. & Cook, J.D. Dose‐finding based on efficacy‐toxicity trade‐offs. Biometrics 60( 3), 684– 693 (2004).
Wiley Online LibraryPubMedWeb of Science®Google Scholar
47Zhang, W., Sargent, D.J. & Mandrekar, S. An adaptive dose‐finding design incorporating both toxicity and efficacy. Stat. Med. 25( 14), 2365– 2383 (2006).
Wiley Online LibraryPubMedWeb of Science®Google Scholar
48Booth, C.M., Calvert, A.H., Giaccone, G., Lobbezoo, M.W., Seymour, L.K. & Eisenhauer, E.A. Endpoints and other considerations in phase I studies of targeted anticancer therapy: recommendations from the task force on Methodology for the Development of Innovative Cancer Therapies (MDICT). Eur. J. Cancer 44( 1), 19– 24 (2008).
CrossrefCASPubMedWeb of Science®Google Scholar
49Francis, M., Basque, J., Legault, E., Rufiange, M., Sicard, E. & Lefebvre, M. An integrated first‐in‐human study design involving sequential single and multiple ascending dose cohorts: an emerging trend in phase I clinical trials. Clin. Pharmacol. Ther. 93, S20– S21 (2013).
Web of Science®Google Scholar
50 Food and Drug Administration, Center for Drug Evaluation Research. In vitro metabolism ‐ and transporter‐mediated drug‐drug interaction studies (draft guidance). https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM581965.pdf (2017) Accessed March 26, 2018.
Google Scholar
51 Food and Drug Administration, Center for Drug Evaluation and Research. Clinical drug interaction studies — study design, data analysis, and clinical implications (draft guidance). https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf (2017). Accessed March 26, 2018.
Google Scholar
52 Immunogenicity assessment for therapeutic protein products. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM338856.pdf (2014). Accessed June 29, 2018.
Google Scholar
53 E14 clinical evaluation of qt/qtc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs ‐ questions and answers (R3) guidance for industry. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073161.pdf (2017). Accessed April 12, 2018.
Google Scholar
54Cutler, N.R. et al. Defining the maximum tolerated dose: investigator, academic, industry and regulatory perspectives. J. Clin. Pharmacol. 37( 9), 767– 783 (1997).
Wiley Online LibraryCASPubMedWeb of Science®Google Scholar
55Cutler, N.R. & Sramek, J.J. Investigator perspective on MTD: practical application of an MTD definition–has it accelerated development? J. Clin. Pharmacol. 40( 11), 1184– 1187 (2000). discussion 1202‐1204.
Wiley Online LibraryCASPubMedWeb of Science®Google Scholar
56 Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for clinical trial sponsors establishment and operation of clinical trial data monitoring committees. https://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm127073.pdf (2006). Accessed April 12, 2018.
Google Scholar
57Goossens, N., Nakagawa, S., Sun, X. & Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 4( 3), 256– 269 (2015).
CASPubMedWeb of Science®Google Scholar
58Arnold, M.E., Booth, B., King, L. & Ray, C. Workshop Report: crystal City VI—bioanalytical method validation for biomarkers. AAPS J. 18( 6), 1366– 1372 (2016).
CrossrefCASPubMedWeb of Science®Google Scholar
59 World Health Organization. The impact of implementation of ICH guidelines in non‐ICH countries. http://apps.who.int/medicinedocs/pdf/h2993e/h2993e.pdf (2001). Accessed April 12, 2018.
Google Scholar
60 Frost & Sullivan. Australia: preferred destination for early phase clinical trials. https://ww2.frost.com/files/6514/7374/3781/Novotech_WP_20160701_v2.1.pdf (2016). Accessed April 12, 2018.
Google Scholar
61Lang, T. A. et al. Clinical research in resource‐limited settings: enhancing research capacity and working together to make trials less complicated. (ed C. H. King). PLoS Negl. Trop. Dis.. 4( 6), e619 (2010).
CrossrefPubMedWeb of Science®Google Scholar
62 International Conference on Harmonisation. Safety pharmacology studies for human pharmaceuticals S7A. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S7A/Step4/S7A_Guideline.pdf (2000). Accessed April 12, 2018.
Google Scholar
63 International Conference on Harmonisation The non‐clinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) S7B. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S7B/Step4/S7B_Guideline.pdf (2005). Accessed April 12, 2018.
Google Scholar
64 International Conference on Harmonisation Note for guidance on toxicokinetics: the assessment of systemic exposure in toxicity studies S3A. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S3A/Step4/S3A_Guideline.pdf (1994). Accessed April 12, 2018.
Google Scholar
65 International Conference on Harmonisation Immunotoxicity studies for human pharmaceuticals S8. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S8/Step4/S8_Guideline.pdf (2005). Accessed April 12, 2018.
Google Scholar
点评:
这是一篇读起来很合我想法的一篇文章~,读起来非常舒服。
- 它非常直接的解答了我在2013~2017年间非常困惑的一些问题——“一个候选药物到底都必须做哪些试验,其中哪些试验又必须是在符合GLP的实验室进行,心脏安全毒性QT间期的试验是哪些药物必须做的”。
- 它十分契合我脑海中关于"首次人体(FIH)“的一些想法,比如“临床操作同事是关键的合作伙伴,可帮助选择研究地点,启动活动,机构审查委员会(IRB)/伦理委员会(EC)批准,合同研究组织(CRO)合作以及研究监督,以确保研究的顺利进行和质量。”,这样的表述才是标准的嘛~,我个人更喜欢把该职能描述为“试验执行者”。
- 它对““首次人体(FIH)“起始剂量的估算”做了一个不错的综述。
- 它对所谓的“试验设计/3+3设计/适应性设计”做了清晰的定位“剂量递增方案”,我个人倾向于认为“试验设计”是一个更大的概念,而“剂量递增方案”只是其中的一个组成部分;它本身也清晰的介绍了“剂量递增方案”的类型与常见设计。
- “安全监察”是一个我不了解的领域,它做了很好的介绍,最近两年“安全监察”是一个很火的概念。
- “生物标志物评估”也是一个很不错的章节,文章叙述介绍了在"首次人体(FIH)“试验中,生物标志物的作用、用途、和鉴定识别依据。
- 结论,这是一篇很棒的文章,它概述了"首次人体(FIH)“研究所需考虑考虑的诸多方面:跨职能合作、适应性研究设计、数据监察、数据收集、监管指南。是一篇良好的综述文章。
文章来源:
本文中的文献原文刊载于Clinical and Translational Science Volume 12,
网址:https://ascpt.onlinelibrary.wiley.com/doi/10.1111/cts.12582
该文是一篇开放访问的可以免费阅读的文章,文章声明适用“CC BY-NC 4.0”协议,可以标注署名的情况下非商业的转载、演绎。