文献:为药物开发中基于临床终点的暴露_反应分析建立的良好实践
付永超 / 2023-05-21
原文链接:https://ascpt.onlinelibrary.wiley.com/doi/10.1002/psp4.12015
原文作者:RV Overgaard, SH Ingwersen, CW Tornøe
译者注:主要有Edge浏览器自带的网页翻译功能翻译,译者额外校对了部分词汇的中文翻译比如“dropout”在本文应翻译为“脱落”,而不是“辍学”
抽象
本教程旨在推广药物开发中基于临床终点的暴露-反应(E-R)分析的良好实践。重点是E-R分析的实际方面,以帮助建模科学家以一致的方式跨个人和项目执行此类分析,并根据典型的临床药物开发决策量身定制。这包括规划、执行和可视化 E-R 分析的一般注意事项,以及这些分析如何与关键问题相关联。
在过去的几十年里,暴露-反应(E-R)分析已成为临床药物开发和监管决策不可或缺的一部分。然而,在更成熟的领域(如群体药代动力学(PK)和药代动力学/药效动力学(PK / PD)建模分析)之后,仍然缺乏E-R的监管指南和行业建议。群体PK分析是一门成熟的学科,关于模型构建、协变量选择和模型诊断的方法论论文众多篇;另一方面,PK / PD建模在后期开始发展,但迅速发展成为一个具有专用期刊(JPKPD)的既定研究领域。PK/PD建模涉及将浓度时间过程(包括给药间隔的变化)与药物动力学反应的时间过程联系起来。
E-R分析在其广义定义中包括PK / PD建模作为特殊情况,其中暴露变量是药物浓度,但大多数情况下,术语E-R是指在几个方面与PK / PD模型不同的分析:I)暴露变量是一个汇总变量,例如曲线下面积(AUC),而不是浓度时间过程。II)反应通常是一个临床终点,通常表示为从基线到试验结束时反应变量的变化。III)安慰剂组的反应和变异性(可能是由于随时间的变化,伴随的药物或安慰剂效应)是分析的核心。IV)在许多情况下,E-R分析是通过简单的回归分析而不是时程模型进行的。在本教程中,我们将关注上述更狭义的 E-R,仅简要提及 PK/PD 和时程建模。
本出版物的目的是为如何在临床药物开发过程中应用E-R分析提供共同的基础。本教程的范围不是讨论理论考虑,而是强调E-R分析的实际方面,以促进个人和项目的一致实施。这包括规划、执行和可视化 E-R 分析的一般注意事项,以及如何将所解决的问题与特定分析联系起来。最后,我们讨论了E-R分析的局限性和假设,以及未来在临床药物开发中的应用前景。重点是分析连续反应数据,但类似的原则适用于分类类型数据。同样,如上所述,我们专注于单个时间点的反应,并且仅就扩展时程E-R分析分享了一些想法,为此仍有待制定标准。
我们希望通过分享我们的观点,定量药理学界将联合起来,开始标准化这些类型的分析,以增加对关键药物开发决策的影响,就像Byon和辉瑞的同事对群体PK分析所做的那样。
暴露-反应分析的良好实践
临床药物开发中E-R分析的主要目的之一是确保在每个开发阶段之后和提交时利用全部可用证据进行充分的剂量选择和证明。为了促进这一点,以下各节提供了与关键问题一致的进行 E-R 分析的一般建议。具体示例将在下一节中介绍。
关键问题
药物开发每个阶段的相关关键问题正被用于将建模支持转向统计推断和定量分析,提供更直接的答案,例如,选择剂量的合理性。表1列出了在患者临床药物开发阶段的设计和解释中需要考虑的关键问题。目的是关注E-R解决的问题,但为了完整性,还包括PK/PD和meta分析解决的问题(例如,基于汇总水平数据,提供给定适应症的试验效果概述)。设计问题通常侧重于试验参数的选择,如剂量方案、试验持续时间和样本量,包括试验提供 E-R 证据的能力,而解释问题侧重于试验数据的 E-R 评估,即确定治疗效果或更高暴露时的更高反应,或旨在表征 E-R 关系。
表 1. 在临床药物开发阶段的设计和解释中需要考虑的通用关键问题
| 阶段 | 设计问题 | 解释问题 |
|---|---|---|
| Ⅰ~Ⅱa | PK/PD分析(例如基于临床前数据)是否支持起始剂量、方案和探索的剂量范围? 模拟是否表明基于I/IIa阶段数据的E-R可以为开发决策提供信息? 该设计是否提供通过生物标志物或临床终点的 E-R 分析检测信号的能力? 设计是否在持续时间、样本量和剂量水平方面进行了优化?考虑其他设计选项是否可以降低错误决策的风险,以及应使用哪些决策标准? 根据对该适应证的荟萃分析,1期生物标志物终点能否可靠地预测晚期终点? | E-R关系是否表明治疗效果? 如果存在安全信号: E-R关系是支持还是挑战与治疗的关系? 在暴露量最高的受试者中,安全问题是否更明显? E-R关系是否表明潜在的治疗窗口? 对于PD标志物,类似的问题可以通过PK / PD分析来解决,PK / PD分析具有更多假设,可能提供比E-R分析更高的功效来检测信号并确定治疗窗口。 |
| Ⅱb | 基于现有数据的PK/PD和E-R分析是否支持建议的剂量范围和方案?IIB 期设计应探索剂量范围,包括亚临床剂量和临床上剂量(如果安全)。 什么是PD时程,包括受试者之间的变异性? 模拟是否表明基于IIb阶段数据的E-R可以为开发决策提供信息? 基于暴露-反应的初级分析/E-R 分析的预测功效是多少? 该设计是否提供检测治疗效果是否随暴露而增加的能力? E-R分析可以帮助确定3期剂量水平吗? 设计是否在持续时间、样本量和剂量水平方面进行了优化?考虑其他设计选项是否可以降低错误决策的风险,以及应使用哪些决策标准? 根据该适应证的荟萃分析,II期终点是否可靠地预测III期结局(如果不同)? | E-R 关系是否支持治疗效果的证据? 功效和主要安全性/耐受性参数的E-R关系的特征是什么? 治疗效果会随着剂量/暴露而增加吗? 什么是最小有效浓度,EC50,最大效果级别?在高曝光时,效果会趋于平稳吗? 预期的治疗剂量/暴露窗口是多少? 对于PD生物标志物:PK / PD关系的特征是什么,包括随时间变化的反应,特别是对于不同治疗方案的试验? 如果存在关键安全信号: E-R关系是否支持或挑战与治疗的关系? |
| Ⅲ和提交 | 基于 II 期数据的 E-R 模拟是否支持 III 期设计、剂量和方案,也适用于具有不同暴露和/或反应的亚群? 初级分析/E-R 分析的预测功效是多少? 第三阶段之后,总体和相关亚组的预期E-R结局是什么?考虑: 人群总体的不确定性? 相关子组在 E-R 曲线上的位置? 如果峰谷比较高:基于 II 期的 PK/PD 模拟是否支持 III 期剂量和方案? 预测的PD时程(包括受试者之间的差异)是多少? meta分析是否表明PD波动对临床终点有影响? | 从组合的2期和3期数据中获得的E-R关系是否支持治疗效果的证据? 功效与主要安全性/耐受性参数的E-R关系的特点是什么?考虑: 治疗效果会随着剂量的增加而增加吗? 什么是最小有效浓度,EC50,最大效果水平,高曝光时效果会趋于平稳吗? 预期的治疗剂量/暴露窗口是多少? 对于关键的安全问题: E-R是否支持或质疑与治疗的关系? E-R分析(总体和亚人群)是否支持拟议的剂量和剂量方案? 剂量变化的预测效果是什么? 与安慰剂相比,是否在所有亚组中都有预期效果? |
表 1 中的问题应被视为通用选项。建议与相关利益攸关方合作,用为每个不同场合量身定制的具体问题来补充或替换这些问题。
设计问题
E-R分析是试验规划阶段的强大工具,可优化设计,以根据化合物和/或药物类别的当前定量信息检测和量化感兴趣的信号。在进行试验之前,应对拟议设计进行模拟和定量探索,以了解设计参数对结果的影响。这包括捕获在特定患者群体中获得预先指定反应的可能性,并进一步探索纳入标准、人口分布、剂量、剂量方案、治疗持续时间和数据分析模型。
仔细规划试验和分析对于在临床药物开发中成功利用E-R分析至关重要。应包括参与试验设计和分析讨论的相关利益相关者,以确保支持和预期分析的重点以及有效利用结果。对于预定义的分析,应在建模分析计划中预先定义分析的详细信息。对于探索性分析,计划应侧重于非技术方面,特别是确定分析要解决的关键问题,以支持内部决策(表1)。
解释问题
解释的关键问题集中在为下一个临床试验的设计或提交给监管机构提供信息。与仅限于为实际临床试验提取证据的经典统计测试相比,E-R和PK / PD分析侧重于通过整合基于药理学和生理学原理的先验知识来得出推论。对剂量-暴露-反应关系的可靠表征可以更好地了解药物的疗效和安全性,并能够做出定量决策,例如剂量选择。一些建议的解释问题与临床开发阶段相关,例如,与治疗效果有关的问题;由于经典统计通常可以令人满意地解决这些问题,因此及时应用E-R分析将避免冗余分析。然而,当统计评估中仍然存在不确定性时,E-R分析可能有助于获得至关重要的证据,例如,从某种意义上说,与暴露的因果关系将增加对观察到的效果的信心。此外,当结果表明需要新的研究时,例如,使用另一种剂量,E-R模型可用于模拟,预测和推断超出观察到的数据,从而用于优化剂量,设计和分析,或完全减轻对进一步临床数据的需求。
数据注意事项
一般来说,研究有能力证明对安慰剂的效果,而不是调查剂量水平之间的差异。因此,如果可能的话,应纳入多项试验进行E-R分析。在IIa期结束时,纳入I期试验的患者数据也是相关的,在提交时,纳入较大的III期和IIb期剂量发现试验也是相关的,这些试验通常跨越更广泛的剂量范围。然而,跨试验的此类分析需要考虑试验设计和研究人群的差异,例如健康受试者与患者,这可能会阻碍有意义的联合分析。此外,早期临床试验的反应数据可能仅限于包括生物标志物而不是临床疗效终点,在这种情况下,分析将依赖于可用的方法将生物标志物数据与临床结果联系起来。
暴露数据可能并非来自所有患者,因此临床试验的 E-R 人群可以定义为完整分析集 (FAS) 中暴露数据可用的患者子集。在主要终点评估时获得的反应数据应与主要分析方法一致的缺失数据进行适当插补;通常混合效应模型重复测量。考虑排除在获得PK稳态之前退出的患者,以降低获得偏倚E-R评估的风险。对于时程分析,所有数据都包括在观察中,并通过混合效应模型进行分析,该模型考虑到如果受试者提前退出,反应可能会降低。单个时间点和时间过程分析的结果将反映所有受试者都继续接受治疗的结果。或者,通过脱落建模更彻底地调查脱落可能是相关的,以提供改变剂量效果的更详细图片。
药物暴露最明显的汇总测量是稳态给药间隔内的浓度-时间曲线下面积(AUC),但其他测量,例如最大药物浓度(Cmax)或谷浓度(Ctrough)可能更合适,取决于适应症、终点和药物的PK性质。在早期药物开发过程中,暴露数据可以通过非间隔方法得出。在后期开发中,稀疏血液采样通常需要使用群体PK分析,在这种情况下,可以使用个体事后AUC估计值作为暴露指标。
反应变量的类型
对于 E-R 分析,反应变量可以是连续、分类或事件时间数据。连续终点的模型通常通过非线性最小二乘过程进行估计。相同类型的模型可用于二进制终点,但数据通常进行逻辑转换以应用非线性逻辑回归,潜在模型如 Emax模型,类似于连续数据的模型。本教程的在线附录中提供了用于分析连续数据和分类数据的模型代码示例。事件时间分析可以在比例风险模型中应用类似的模型,或使用模型独立分析,例如不同暴露分位数或安慰剂的受试者的 Kaplan-Meier 图。
假设和限制
E-R 分析与假设和限制相关联,在收集数据、进行分析或解释结果之前应考虑这些假设和限制,如下所示。
假设
-
暴露变量(可能由模型推导)准确反映了个体平均有效浓度:即E-R分析与剂量无关。这可以通过比较不同剂量水平的E-R关系来图形化方式地解决。
-
排除没有充分暴露测量的受试者不会使结果产生偏差。这可以通过比较有和没有足够暴露测量的受试者随时间推移的反应来解决。
-
假设对疗效终点使用插补不会偏倚结果。由于受试者脱落的真实结果是未知的,任何插补都可能产生有偏见的结果。对于后期试验,主要分析通常需要进行敏感性分析以解决此问题。
-
分析中考虑了所有相关的混杂协变量(请参阅下面的分析方法选择)。这可以通过检查测试剂量水平的E-R模型预测与观察到的剂量 - 反应关系之间的一致性来探索,因为如果患者在剂量水平之间随机分配,则剂量 - 反应关系不应存在混杂协变量,例如,某个亚组的较高反应和剂量水平。但是,一般来说,不可能完全验证这一假设。
-
参数模型充分表示真实 E-R 关系的形状。这可以通过拟合优度图进行评估。
-
EC50和Hill 系数(见下一节)在贡献试验中或人群之间被假设相同。这些参数的任何差异都不太可能与Emax的差异同时被识别,因为数据可能不足。但是,如果数据不足以确定人群之间的差异,则违反这一假设不太可能产生重大影响。
局限性
-
E-R分析通常基于不平衡的数据,因为例如,重的受试者可能与轻量级受试者具有不同的暴露,这可能会限制统计测试方面的应用。在暴露方面缺乏平衡是一个固有的限制,因为临床试验通常是在剂量方面控制的,而暴露是一个不受控制的变量。一些科学家发现这是一个关键的局限性,导致对E-R分析的怀疑,但我们需要强调的是,大多数情况下,缺乏随机化不是问题,这些分析的优势应该鼓励E-R分析的一般应用,增加决策证据的整体。
-
E-R分析最重要的限制是可能存在无法识别的混杂因素。这将在分析方法的选择一节中进一步讨论。
-
E-R分析的目的是分析特定终点的反应,即在给定的时间点,这不一定反映药物的长期影响。
-
上述假设和局限性不大可能使分析无效,除非出现很高的数据排除率。对于后期应用尤其如此,其中暴露和反应的重要协变量可能已被确定,并且已考虑混杂因素。
分析方法的选择
用于进行E-R分析的方法应根据正在解决的问题仔细调整。
从根本上说,我们正在根据问题使用三种不同类型的分析来解决E-R问题,如表2所示。第一个问题涉及相对于安慰剂的效果,而后两个分析阐明了与选择和支持剂量相关的各个方面。
表 2. 通用关键问题,以及用于解决问题的建议模型
| 类型 | 问题 | 分析* |
|---|---|---|
| A | 数据是否可以表明治疗效果? | ECFB∼ EBASE + COVs + Slope×Exposure+Intercept (E主要终点相对与基线变化~E基线+协变量+斜率×暴露量+截距) (基于所有数据的分析) |
| B | 治疗效果是否会随着剂量的增加而增加? | ECFB∼ EBASE + COVs + Slope×Exposure+Intercept (E主要终点相对与基线变化~E基线+协变量+斜率×暴露量+截距) (不包括安慰剂数据) |
| C | E-R关系的特点是什么? 剂量变化的预测效果是什么? | ECFB∼ EBASE +COVs +Emax ×Exposure /(EC50 + Exposure) +Intercept (E主要终点相对与基线变化~E基线+协变量+Emax×暴露量/(EC50+暴露量)+截距) (基于所有数据的分析) |
*ECFB表示与主要终点相对基线的变化, EBASE的是效应变量的基线值。Exposure是一个暴露变量,例如稳态给药间隔内浓度-时间曲线下的面积。COV 是协变量对效应的贡献。Slope是线性尺度上 E-R 关系的估计斜率。该 Emax模型(C型)由Emax和EC50参数化,Emax是在无限暴露时获得的最大效应,EC50是在半数最大效应时的暴露。对于任何分析,都包括一个截距,代表零暴露(即安慰剂)下的反应。方程以ECFB为因变量,假设为连续型终点。类似的分析可以应用于分类型的二分类终点,在 logit 转换之后,并使用反应率作为因变量。
前两种类型的分析(A 型和 B 型)应该是预定义的或相对通用的,以避免多次检验,以确保可解释的 P 值。表征和预测的第三个分析(C型)意味着开始模型开发,以获得结构关系和协变量效应的最佳描述。正如数据所暗示的那样,结构模型可以简化为(对数)线性关系或用Hill系数扩展,并且应为相关参数选择协变量,以获得最佳和最真实的描述。这些根本不同类型的分析可能有助于回答整个临床药物开发过程中的不同问题,如下所述。
对于任何关键问题,我们建议应用最简单的分析,其中包括最少的假设,并且可以很容易地传达给广泛的利益相关者。因此,建议将上述简单的单时间点分析作为确定的起点。然而,这三类分析构成了E-R的充分工具箱,这是一种粗略的简化,因为许多方面尚未得到解决。特别是,使用整个数据时间过程的脱落模型和混合效应分析可以提供有价值的额外见解。这些更先进的技术的应用通常是探索性的,是基于数据而不是预定义的。应根据具体情况考虑一些潜在的模型扩展,以考虑脱落、耐受性发展和疾病进展。有关这些选项的全面建议不在本教程的讨论范围之内。相反,我们的建议集中在使用相对简单的模型来评估单个时间点的暴露与治疗结果之间的关系,我们认为这对于药物开发中的大多数情况是足够的,并且最接近协议中指定的主要分析。但是,以下各节提供了一般性说明,以指导时程 E-R 分析和需要考虑脱落的情况。
时程 E-R 模型
原则上,反应数据时程模型构成了 E-R 分析的标志,它以牺牲更多假设为代价,提供了比单个时间点分析更多的见解。特别是,如果反应不是稳定状态,例如,由于耐受性的发展或疾病的进展,时间序列模型可以提供与现实世界使用高度相关的长期预测。然而,对于某个时间点的临床终点调查,单个时间点分析通常就足够了。一般而言,我们建议在以下情况下使用时程分析来补充表 2 中概述的单个时间点分析:
-
反应的场合间变异性很大。
-
主要终点处的反应似乎与反应的整个时间过程不一致。
-
在频繁的或信息性的脱落情况下。
对于E-R分析,安慰剂组的反应是需要考虑的重要因素。同样,时程模型需要包括随时间变化的安慰剂成分。一个经常应用的安慰剂模型是:
$$ E_{placebo} =E_{max,pl}*(1-e^{-k_{pt}*time}) $$其中 Emax,PL是最大安慰剂效应和kpl是安慰剂效应发展的速率常数。活性组的总反应可以表示为安慰剂效应和治疗效果的总和。
尽管 E-R 分析通常基于汇总暴露统计量(例如 AUC)而不是完整的 PK 时间过程,但 E-R 模型可能会借用经典 PK/PD 建模(如间接反应模型)中的方法来描述治疗效果随时间的变化,同时考虑到剂量是否随时间变化。然而,一个简单的替代方法是仅使用一个稳态暴露值,以及与上述安慰剂效应类似的模型,例如:
$$ E_{treatment} =\frac{E_{max} *Exposure_{ss}}{(Exposure_{ss }+EC50)} *(1-e^{-k_{TR}*time}) $$其中$Exposure_{ss}$是一个稳态暴露变量,例如 AUC,$k_{TR}$是完全治疗效果发展的速率常数,并且 Emax是最大反应。
这种设置的一个问题是,潜在的安慰剂效应和治疗效应加在一起,安慰剂效应和治疗效果并不总是分开的,即,如果受试者的反应超过平均水平,这可能是由于安慰剂反应升高或治疗效果升高。在单时间点分析中,人们通常通过安慰剂和积极治疗中具有相似方差的单个残差来捕获这种升高的反应。对于时程分析,相应的方法不是包括治疗效果的随机个体间变异性,而只是安慰剂效应。虽然这将是一个有吸引力的起点,但它可能不是最佳选择,应根据具体情况进行探讨。
脱落模型
如果忽略患者脱落,单个时间点和时间过程分析的结果将反映如果所有受试者都继续接受治疗时将获得的反应。如果大量受试者退出试验,则可能需要通过在模型中包括脱落成分来更彻底地调查脱落,以便提供更现实的评估。因此,为了预测改变剂量的效果,预测哪些患者实际上会完成试验是相关的。这允许预测完成者以及退出试验的受试者的反应。此外,脱落模式(所有脱落,由于缺乏疗效或不良事件而脱落)本身可以被视为临床终点,因此与E-R分析相关。
脱落模型是事件发生时间分析,通常基于比例风险模型的原则,并扩展取决于浓度/暴露、副作用水平和疗效的术语。实施脱落模式的灵感,特别是信息丰富的脱落,可以在其他地方获得。
在本教程中,我们主张简单,不建议在 E-R 分析中通用实现脱落模型。事实上,如果患者坚持治疗(如忽略脱落的模型提供),而不是旨在预测意向性治疗人群的真实结局,评估预期结局通常可能信息量最大且合理充分。然而,对于超过20%的脱落率,我们建议将其作为独立的临床终点进行研究,特别是与不良事件相关的脱落。此类评估应包括在益处和风险的评估中,例如,在预测不同给药方案的结果时。
协变量和子组的评估
协变量对于评估患者亚组中的 E-R 关系和调整混杂因素(同时影响 PK 和反应变量的协变量)非常重要。如果没有正确考虑,已知和未知的混杂因素可能会影响分析,这可能是E-R分析中最重要的误差来源。图1说明了在模型中将性别作为协变量包括在内的重要性,以解释女性比男性更高的暴露。从图 1a 可以看出,Emax具有Hill系数的模型表示对数据的良好拟合。然而,通过包括混杂协变量(性别),很明显真正的模型(一个简单的Emax关系)更合适,现在证明在整个暴露范围内反应几乎是恒定的,如图1b所示。
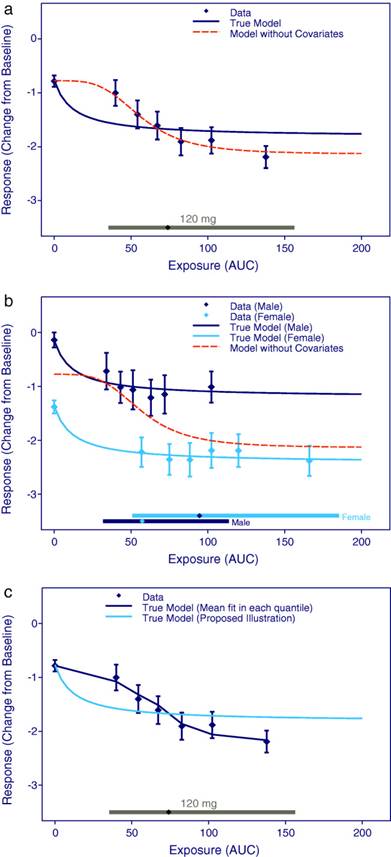
图1
可视化 E-R 关系 (a) 没有混杂协变量(性别)的分层 (b) 有混杂协变量(性别)的分层。(c)对于每个分位数,所提出的模型可视化方法与模型拟合进行了比较。数据点是 AUC 值分位数的平均效应,置信区间为 95%,线表示估计的 E-R 关系。沿着横坐标的菱形水平线代表每个剂量水平的中位数和 90% 暴露范围。这三个面板基于模拟1,000名男性和女性平均分布的受试者生成的相同数据集。
因此,分析需要考虑可能的混杂因素,将这些因素作为协变量包括在内。最近发表了作为敏感性分析的病例对照调整,作为在独立于模型的分析中调整已知混杂因素的一种方法。
通常,建议根据临界程度限制协变量的数量。应考虑以下协变量:
始终包括基线时的反应变量和已知与PK和/或反应相关的协变量。对其他协变量的调查可能是相关的,例如:
-
试验/人群/附加治疗。
-
年龄、体重、性别、世界地区。
-
其他人口统计数据(例如,其他非PK相关协变量),相关的伴随药物/疾病等。
协变量的实现可能很棘手,因为太多参数上的太多协变量会增加不确定性,并可能使参数估计无法进行。一般来说,对于高精度估计的参数,可以包括更多的协变量,例如,与更不确定的参数,如Emax或EC50相比,对积极治疗和安慰剂具有相同的影响基线效应的协变量(见下文)。
基线效应的协变量(对积极治疗和安慰剂的影响相似)。可以毫无问题地包含多个协变量,但应保留相关和不相关的协变量:
-
对于具有大量患者的III期数据:包括上面列出的最重要的协变量。仅在需要时才考虑其他协变量。
-
如果患者数量有限,例如IIa期:仅将反应的基线值作为协变量包括在内。对于已知的混杂协变量(或预先指定的分析),例如,如果性别与PK和反应有关,则分别对男性和女性进行额外的分析。
治疗效果的协变量:
-
应研究这些协变量,最好是 Emax最终模型中只应包含少数(例如,0-2)协变量,以避免过度参数化。
-
不要在E-R(A型分析)证据的统计检验中包括此类协变量。如果相关,分别检测患者的亚组。
-
不要为 EC50 实现协变量除非个体受试者有不同剂量水平的数据,或者从数据中可以明显看出这种协变量。EC50上的协变量通常可以用Emax上的协变量代替。
结果可视化
E-R分析可以作为图形数据分析和/或基于模型的分析进行。图形数据分析通常在启动基于模型的分析之前执行,并在可能的情况下对基于模型的分析进行限定。有时,图形分析可能是特定任务的唯一选择方法。
观测值的可视化
由于反应的可变性,作为个体反应与暴露的散点图的图形分析通常变得复杂且难以解释。分类终点当然是这种情况,但大多数情况下也是连续设置。相反,我们建议将受试者划分为暴露分位数,如下所示。对于图形图中的每个亚组/试验,积极治疗的受试者根据其暴露值分为分位数。对于每个分位数(或安慰剂),将反应的平均值和95%置信区间(CI)与中位暴露水平作图,用安慰剂治疗的受试者的暴露值为0。对于在小数据集上进行的多个分析,对所有分析使用相同的暴露分位数可能是有利的,以便更好地将一个时间点/终点与另一个时间点/终点进行比较。在其他情况下,确保所有分位数中包含相同或相似数量的受试者可能更为重要。这种分位数图通常提供数据趋势的有用信息,因此也可用于鉴定基于模型的预测。
模型派生预测的可视化
基于模型的分析的一个关键目的是考虑到暴露程度最高最高的受试者比暴露量最低的受试者更有可能具有不同的人口构成。为了解决这些问题,建议通过以下过程生成平均暴露-反应模型预测:
-
对于数据集中的每个受试者,获取实际的协变量(性别、体重等),并使用模型预测整个暴露范围内对许多离散预先固定的暴露值的反应。
-
在这些暴露值每一处,计算每个试验/亚组中所有受试者的平均模型预测。
此过程可确保模型预测的整个暴露范围内的反应差异由暴露差异驱动,而不是由整个暴露范围内的协变量分布的变化驱动。在本教程中,所有模型和图形都使用了所提出的模型结果呈现方法。图1c通过将所提出的方法与为每个分位数暴露获得的平均模型拟合进行比较,说明了该方法的重要性。如图所示,所提出的模型可视化方法说明了真正的E-R关系是平坦的,因此除了简单的数据说明之外,还提供了其他信息。另一方面,如果我们选择在每个分位数中呈现均值模型拟合(这可能是一个相关的模型诊断图),我们可能会错误地得出结论,即模型支持更高的暴露提供更高的反应。
通常,E-R的可视化包括不同的扩展,例如,受试者之间的变异性或预测中的不确定性。包含预测不确定性可能是相关的,例如,对于统计结论,或者在分析和比较来自几个不同终点的收益和风险时。然而,模型不确定性的一般引入可能不太相关,因为不确定性可能在很大程度上取决于假设、参数化和复杂性。例如,如果建模者包含的分量不随剂量单调增加,则这些分量可以很容易地提供类似的均值 E-R 关系,但不确定性估计值会发生较大变化。本教程中介绍的分位数周围的置信区间提供了对不确定性的模型独立评估。
可视化反应的时间过程
时程模型的诊断图通常包括反应数据与时间的关系,并叠加模型预测。为了使时程模型的应用侧重于暴露的影响,我们还建议包括反应与暴露的分位数图,例如,在试验结束时进行模型预测,总体上具有相似的分析,以及亚组(类似于上述单时间点分析的建议)。在不采用时程模型的情况下,我们建议包括暴露/剂量亚组的反应与时间的探索性图表,以调查单个终点分析是否足够。
示例:链接关键问题和分析
表2介绍了关键问题与相应的E-R分析之间的联系。以下部分将更详细地解释何时以及为何应用不同类型的分析。
数据是否表明治疗效果?(A类问题)
在早期开发过程中,最重要的问题是该化合物是否有效,即在I/IIa期之后建立临床原理证明,以决定是否进行全面的临床药物开发。由于现阶段提供数据的患者数量有限,因此在每次剂量使用经典的治疗结果统计测试与安慰剂相比时,功效可能有限。出于这个原因,使用所有可用剂量数据的E-R分析可以提供重要的支持有效性证据。
尽管真正的 E-R 关系可能更复杂,但为此,建议使用预先指定的模型使用线性近似进行通用分析(参见表 2 中的类型 A)。如图2所示,I/IIa期试验数据的变异性可能排除了关于E-R关系形状的任何结论,但是,可以使用线性关系的假设和测试线的斜率是否与零显着不同来确定治疗效果。
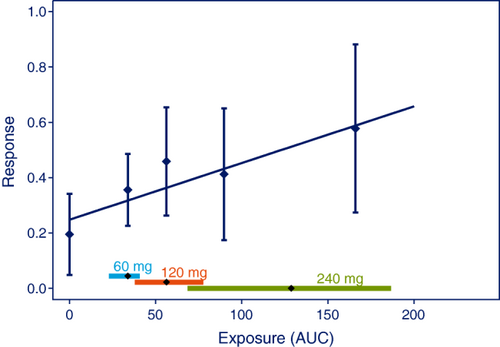
图2
绘制以可视化预先指定的分析以建立有效性的支持性证据的绘图。数据点是 AUC 值分位数的效应的平均值和效应的95% CI。沿着横坐标的菱形水平线代表每个剂量水平的暴露量的中位数和暴露量的90%暴露范围。在两个较低剂量水平下模拟8名活性+4名安慰剂受试者的数据,在最高剂量水平下模拟16名活性+8名安慰剂受试者的数据。
显然,将治疗效果确定为具有统计学意义不仅在I/IIa期结束时,而且在整个开发过程中的许多问题中都具有相关性。这可能与III期的主要终点无关,但在其他情况下,E-R分析可能有助于得出临界效应是否真实的结论。例如,对于安全性数据,将E-R分析作为整体证据的一部分进行非常重要,以支持或质疑使用经典统计分析看起来不清楚或可疑的发现。
药物效应会随着剂量的增加而增加吗?(B类问题)
对于已经确定的效果,通常与测试效果是否在整个研究剂量范围内增加有关,以评估一种剂量水平相对于另一种剂量水平的益处和风险。当使用E-R分析从一个剂量水平过渡到另一个剂量水平时,这个问题通常适用,例如,通过说明安全生物标志物在所有剂量水平上的反应相似,如图3所示。在提交阶段,当试图对不同的终点得出可靠的(是/否)结论以证明推荐剂量的合理性时,这个问题可能特别有用。在药物开发的早期阶段,例如,在设计III期试验时,剂量选择可以基于预期的E-R关系,即使用C型分析(表2)来确保III期剂量水平将提供足够的疗效和安全性,即使在特别高或低暴露的受试者中也是如此。
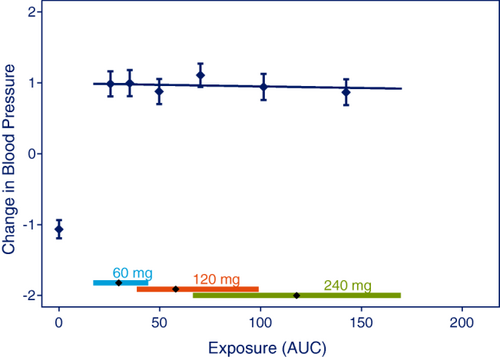
图3
用于可视化用于测试已建立的效果是否在研究剂量范围内随剂量增加的分析的图。数据点是 AUC 值分位数的平均值和 95% CI 效应。沿着横坐标的菱形垂直线代表每个剂量水平的中位数和 90% 暴露范围。对240名受试者在每个剂量水平或安慰剂下模拟数据。
与 A 型问题类似,B 型问题(表 2)可以通过使用预先指定的效应与暴露线性模型为斜率建立 P 值来解决。然而,在该分析中,安慰剂数据必须从分析中排除,以确保安慰剂组不会得出假阳性结论。如图3所示,与安慰剂数据相比,治疗效果可能很明显,但是当仅考虑研究的剂量范围并在分析中忽略安慰剂效应时,可能不存在E-R关系。
E-R关系的特点是什么?(C类问题)
基于主要疗效终点的E-R关系模型以支持建议的剂量水平可能是最常见的E-R分析类型。与早期临床药物开发相比,IIb/III期更是如此,后者提供的信息较少。III期通常以一到两个剂量进行,如图4所示,为了覆盖足够大的剂量范围,为该分析包含II期数据可能至关重要。
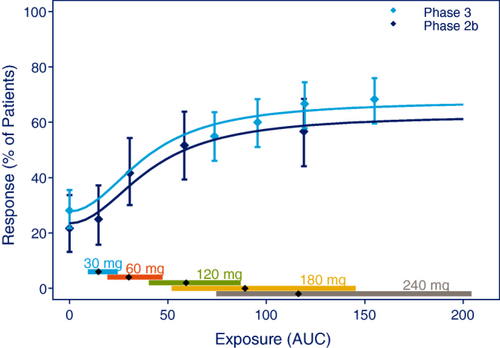 图4
图4
从II期和III期试验中获得的E-R关系。数据点是 AUC 值分位数的平均效应,置信区间为 95%,线表示估计的 E-R 关系。沿着横坐标的菱形垂直线代表每个剂量水平的中位数和 90% 暴露范围。数据是一项模拟的IIb期试验,包括60名受试者,每种剂量水平为30、60、120、240毫克或安慰剂,以及一项模拟的III期试验,将160:160:320受试者随机分配到安慰剂、180毫克或240毫克。
虽然这些彻底的C型分析目前在疗效终点中更为常见,但它们对于安全性或耐受性终点同样重要,以便证明不同剂量水平的获益-风险,例如,在表3等表格中总结,对于整个人群和选定的亚群。这可能涉及探索替代剂量,例如,说明较低剂量将提供不充分的效果,并且较高剂量将与有限的额外益处或不可接受的耐受性相关联。为了提供准确的数据描述和最佳预测,通常需要使结构模型和协变量适应实际数据。虽然这种类型的分析提供了对剂量变化的预期影响的最准确预测,但B型分析(表2)对于提供确切的结论更有用,例如两个最高剂量水平的影响之间是否存在差异。
表 3. 假设剂量从120mg增加到180mg的预测反应
| 剂量增加120毫克→180毫克 | 预测效果增加 | 有耐受性问题的受试者百分比的预测增加 |
|---|---|---|
| 所有受试者 | 5% | 7% |
| 10% 最重的受试者 | 8% | 9% |
| 10% 最轻的受试者 | 3% | 5% |
除了这种B型分析,将一种剂量与另一种剂量进行比较外,通常还应用C型分析来表征整个剂量/暴露-反应关系,以证实剂量是足够的,在治疗范围内提供暴露。在进入III阶段之前,应用这种整体E-R表征来说明极端暴露受试者的预期结果,并确保所包含的至少一个剂量水平有望提供足够的安全性和有效性。对于整个人群和相关的患者亚组,都应对提交材料进行类似的分析。
讨论
在过去的十年中,E-R分析已经发展成为一门独立的学科,在此期间,科学文献中提出了许多具体案例。
E-R 分析建议摘要
本教程强调了在整个临床药物开发过程中应用E-R分析的重要性,作为理解疗效和/或安全性数据的工具,以及作为决策的定量支持。表 4 概述了主要建议。
表 4. 本教程中提供的主要建议摘要
| 关键问题 | • 应用关键问题,与利益相关者保持一致,作为建模分析计划的核心部分。 • 用针对每种研究药物的定制问题补充一般性问题。 • 考虑传统的PK/PD模型还是E-R模型最适合解决问题。 |
| 数据注意事项 | •指定相关的数据集和数据插补,以便对暴露反应结果进行有用的解释,而不会对结果产生明显的偏差。 •对于中高脱落率,脱落可能与作为独立临床终点的研究相关。 • 论证暴露变量的选择(AUC是一种常见的选择,但并不总是合适的)。 • 在有意义的情况下,将PK取样纳入后期临床药物开发。 |
| 假设和限制 | • 尽可能解决假设问题,例如通过诊断图和敏感性分析。 •由于可能存在未知的混杂因素,E-R分析通常是支持性证据,而不是主要分析。 |
| 分析方法的选择 | • 使分析与关键问题保持一致。 • 考虑模型是否应预先指定,还是基于数据的模型开发/选择是否更合适。 • 始终估计单个时间点模型,并可能通过探索性图调查更高级的模型是否相关。 由于高场合间变异性或由于脱落导致的偏差,时程 E-R 模型可能具有相关性。 脱落建模也可能与在研究完成者总体或使用特定插补策略的ITT分析时提供更细微的反应图有关。 |
| 协变量 | • 协变量很重要,以便调整混杂因素。 • 对于精度较高的参数,可以包含更多的协变量。 基线效应可以包括几个协变量(相关性有限);在预先指定的模型中也是如此。 限制Emax的协变量数量(通过向前纳入和向后剔除来减少)。 只有当数据明显且预测在生理上合理时,才包括EC50和Hill系数的协变量。 |
| 结果可视化 | • 通过分位数图可视化结果,例如显示反应的平均和 95% CI 与中位暴露的关系。 • 始终添加考虑已知混杂因素的模型预测。 • 模型预测应反映所有暴露水平下的所有受试者。 • 考虑预测区间或置信区间是否提供了相关且客观的附加信息。 |
非常重要的一点是,我们建议将具体的E-R分析与关键问题联系起来,以便提供清晰的结果并促进与内部和外部利益相关者(包括监管机构)的沟通。出于沟通目的,重要的是将暴露和反应之间的关系显示为模型衍生的估计值,并覆盖了数据。为此目的使用单个数据点通常会模糊数据的趋势,因此我们建议使用暴露值的分位数来显示观察到的数据。可以调整分位数,以便以适当的分辨率显示数据。
使用基于模型的E-R分析通常基于应定义并传达给利益相关者的假设。原则上,可能无法验证所有假设,因此可以考虑使用纯数据摘要作为基于模型的分析的替代方案。但是,使用基于模型的分析,可以通过在模型中包含适当的协变量来研究混杂因素的存在。通过图形数据分析中的分层,这并不总是可行的。此外,基于模型的E-R分析在调查患者亚组和探索剂量方面提供了额外的机会,这些机会尚未在试验中进行测试。由于这些原因,我们认为基于模型的分析是首选方法,除非出现反对它的证据。
E-R分析在临床药物开发中的应用
一开始,E-R分析主要作为后期临床药物开发的工具进行探索。稀疏PK采样和群体PK分析在验证性临床试验中的广泛使用,为根据AUC值提供药物暴露的个体稳态估计提供了可能性,这适用于E-R分析。从那时起,许多应用已经出现,涵盖了从早期到后期临床药物开发的整个链条,如本教程所述。
通常,E-R分析旨在量化特定暴露范围内的影响,而不是提供效果的统计证明。因此,E-R分析通常被视为有效性的支持性证据,补充了试验终点的统计评估,因为基础数据通常是不平衡的,模型假设并不总是得到验证。
然而,使用基于模型的临床试验评估已被证明可以显着提高功效,时间将证明这种方法在未来临床试验中作为主要分析工具是否具有越来越重要的作用。
另一方面,在本教程中,我们已经表明E-R分析具有建立早期临床原理证明的明显潜力。这可以通过对暴露与反应的线性近似中的显着斜率进行统计验证来实现。这种确认性申请对于决定使用候选药物进入全面临床药物开发可能很有价值。
目前,E-R分析在临床药物开发中的应用可总结如下:
-
建立临床原理证明。
-
提供推荐剂量的理由。
-
亚人群推荐剂量的理由;
-
将结果从一个剂量水平与另一个剂量水平联系起来;
-
探索假设的剂量变化。
-
-
对意外发现的解释。
-
提供有效性的支持性证据。
-
探索不良反应是否与药物暴露有关。
观点
E-R分析在许多情况下的成功应用提出了一个问题,即未来是否可能出现新的和更广泛的应用。一种可能的发展是使用基于模型的E-R分析来评估试验结果,作为经典统计评价的替代或补充。如上所述,研究表明,与使用基于模型的 E-R 分析作为主要试验分析的统计测试相比,功效更高。使用此类工具评估帕金森病和高血压的试验结果的例子是存在的。基于模型的治疗结果评估的固有局限性和假设是否会限制这一特定应用的发展还有待观察。
为了便于E-R分析,建议在临床后期药物开发中包括PK采样,如果有意义。我们已经体验到,这种分析有助于更深入地了解支持推荐剂量的数据,尤其是为了解决有效性和安全性方面的监管问题。然而,我们确实承认,对于某些项目,广泛的PK采样是没有意义的,例如,对于剂量滴定以达到效果的药物。
E-R模型可用于估计关键试验中未包括的剂量的试验结果,并且存在基于E-R批准此类剂量的情况。时间将证明,对E-R分析的更多关注是否会刺激更多的此类应用,从而在降低成本和时间方面提供有吸引力的机会。
鉴于E-R分析在药物审批中的广泛使用,不仅用于疗效,还用于安全性评估,因此出现了一个问题,即基于模型的分析是否会发展成为获益风险评估的标准工具。已经看到朝着这种情况发展的趋势,但需要“消除”许多障碍才能进一步继续这一发展。
制定准则和良好做法
在过去十年中,E-R分析的使用扩大部分是由监管举措推动的,例如美国食品和药物管理局(FDA)的2a期结束会议和基于问题的临床药理学审查,其中包括几个E-R问题。
FDA和欧洲药品管理局(EMA)提供了E-R分析的监管指南。据信,修订后的指南提出了更具体的建议,可能会进一步刺激申办者一致使用E-R分析。这些指南将受益于以下具体建议:I)在后期临床试验中收集PK样本;II)建立治疗窗口的期望;III)对整个患者群体以及患者亚组进行评估,包括分析具有极端人口统计学特征(如低体重和高体重)的患者;IV)用于评估临床试验中未专门测试的剂量。朝这个方向迈出的一步是当前的EMA/EFPIA倡议,即制定良好实践指南,旨在提高模型知情药物发现和开发(MID3)的一致性、质量和透明度。
本良好实践文件首次尝试分享在临床药物开发中进行E-R分析的行业良好实践。作者鼓励其他公司分享他们的内部建议,以促进关于如何标准化这些类型的分析以最大限度地影响临床药物开发决策的知情讨论。
参考文献
标题翻译版
· 1 Pinheiro, J. & Duffull, S. 暴露反应-选择正确的剂量. Pharmaceut. Statist. 8, 173– 175 (2009).
· 2 Ette, E.I. & Williams, P.J. 群体药代动力学I:背景、概念和模型. Ann. Pharmacother. 38, 1702– 1706 (2004).
· 3 Ette, E.I. & Williams, P.J. 群体药代动力学II:估计方法. Ann. Pharmacother. 38, 1907– 1915 (2004).
· 4 Ette, E.I. & Williams, P.J. 群体药代动力学III:群体药代动力学研究的设计、分析和应用. Ann. Pharmacother. 38, 2136– 2144 (2004).
· 5 Sherwin, C.M.T., Kiang, T.K.L., Spigarelli, M.G. & Ensom, M.H.H. 群体药代动力学建模基础,验证方法. Clin. Pharmacokinet. 51, 573– 590 (2012).
· 6 Bonate, P.L. et al. 群体药代动力学-药效动力学分析质量控制指南:行业视角. AAPS J. 14, 749– 758 (2012).
· 7 Meibohm, B. & Derendorf, H. 药代动力学/药效动力学(PD/PD)建模的基本概念. Int. J. Pharmacol. Ther. 35, 401– 413 (1997).
· 8 Csajka, C. & Verotta, D. 药代动力学-药效动力学建模:历史与展望. J. Pharmacokin. Pharmacodyn. 33, 227– 229 (2006).
· 9 Byon, W. et al. 建立群体建模的最佳实践和指南:内部群体药代动力学分析指南的经验. CPT Pharmacometrics Syst. Pharmacol. 2, e51 (2013).
· 10确认性试验中缺失数据的指南, EMA (2011). <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/09/WC50 0096793.pdf>
· 11 O’Neill, R.T. & Temple, R. 临床试验中缺失数据的预防和治疗:FDA对处理缺失数据重要性的看法. Clin. Pharmacol. Ther. 91, 550– 554 (2012).
· 12 临床试验中缺失数据的预防和治疗; National Research Council. The Prevention and Treatment of Missing Data in Clinical Trials (National Academies Press, Washington, DC, 2010). <http://www.nap.edu/catalog.php?record id=12955#orgs>
· 13 Holford, N. 为药理学家提供的活动时间指南. CPT Pharmacometrics Syst. Pharmacol. 2, e43 (2013).
· 14 Hu, C. 利用潜变量间接反应模型对临床终点的暴露-反应建模. CPT Pharmacometrics Syst. Pharmacol. 3, e117 (2014).
· 15 Björnsson, M.A., Friberg, L.E. & Simonsson, U.S. 在存在信息性脱落的情况下执行非线性混合效应模型. AAPS J. 17, 245– 255 (2015).
· 16 Hu, C. & Sale, M.E. 具有信息脱落的非线性纵向数据的联合模型. J. Pharmacokinet. Pharmacodyn. 30, 83– 103 (2003).
· 17 Yang, J. et al. 监管决策中暴露-反应分析和病例对照分析的结合. J. Clin. Pharmacol. 53, 160– 166 (2012)
· 18 Goyal, N. & Gomeni, R. 抗抑郁药物治疗的暴露-反应模型:安慰剂效应的混淆作用. J. Pharmacokinet. Pharmacodyn. 40, 389– 399 (2013).
· 19 Hu, C., Wasfi, Y., Zhuang, Y. & Zhou, H. 在暴露-反应模型中的荟萃分析贡献的信息:在中到重度银屑病患者中应用Guselkumab的第二阶段剂量选择. J. Pharmacokinet. Pharmacodyn. 41, 239– 250 (2014).
· 20 Bouazza, N. et al. 儿童HIV-1感染患者洛匹那韦/利托那韦的量效模型. Pediatr. Infect. Dis. J. 33, e213– e218 (2014).
· 21 Pouw, M.F. et al. 优化阿达利单抗治疗的关键发现:浓度-效应曲线. Ann. Rheum. Dis. 74, 513– 518 (2015).
· 22 Wang, Y. et al. 晚期试验中的PK. Appl. Clin. Trials 21, (2012).
· 23 Karlsson, K.E., Vong, C., Bergstrand, M., Jonsson, E.N. & Karlsson M.O. 概念验证试验分析方法的比较. CPT Pharmacometrics Syst. Pharmacol. 2, e23 (2013).
· 24 Bhattaram, V., Siddiqui, O., Kapcala, L.P. & Gobburu, J.V.S. 终点和分析,以辨别早期帕金森病的疾病修饰药物效果. AAPS J. 11, 456– 464 (2009).
· 25 Lee, J.Y. 定量药物学分析对新药批准和药品说明书决定的影响:对2000至2008年间提交的198份材料进行审查. Clin. Pharmacokinet. 50, 627– 635 (2011).
· 26 Overgaard, R.V. 暴露-反应-剂量它为MBDD提供了更多内容,以及如何让我们到达那里, Las Vegas (2014).
· 27行业指南,临床试验2A阶段结束会议. Center for Drug Evaluation and Research, FDA (2009). <http://www.fda.gov/downloads/Drugs/…/Guidances/ucm079690.pdf>
· 28临床药理学和生物药剂学(CPB)审查模板:基于问题的审查(QBR). Center for Drug Evaluation and Research, FDA (2004). http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/StaffPoliciesandProcedures/ucm073007.pdf
· 29良好审评做法:对研究性新药申请进行临床审评. Office of New Drugs in the Center for Drug Evaluation and Research, FDA (2013). http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/UCM377108.pdf
· 30行业指南、暴露-反应关系–研究设计、数据分析和监管应用. Center for Drug Evaluation and Research, FDA (2003). http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072109.pdf
· 31关于药物开发有效性和安全性推断的概念性文件. EMA (2013). <http://www.ema.europa.eu/docs/en_GB/document_library/Other/2013/04/WC500142359.pdf http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/04/WC500142358.pdf>
· 32 Milligan, P. et al. 示范知情药物发现和开发的良好做法(MID3):实践、应用、文件和报告. Poster presented at the American Conference on Pharmacometrics, Las Vegas, 2014.
英文原版
· 1 Pinheiro, J. & Duffull, S. Exposure response—getting the dose right. Pharmaceut. Statist. 8, 173– 175 (2009).
· 2 Ette, E.I. & Williams, P.J. Population pharmacokinetics I: background, concepts, and models. Ann. Pharmacother. 38, 1702– 1706 (2004).
· 3 Ette, E.I. & Williams, P.J. Population pharmacokinetics II: estimation methods. Ann. Pharmacother. 38, 1907– 1915 (2004).
· 4 Ette, E.I. & Williams, P.J. Population pharmacokinetics III: design, analysis, and application of population pharmacokinetic studies. Ann. Pharmacother. 38, 2136– 2144 (2004).
· 5 Sherwin, C.M.T., Kiang, T.K.L., Spigarelli, M.G. & Ensom, M.H.H. Fundamentals of population pharmacokinetic modeling, validation methods. Clin. Pharmacokinet. 51, 573– 590 (2012).
· 6 Bonate, P.L. et al. Guidelines for the quality control of population pharmacokinetic-pharmacodynamic analyses: an industry perspective. AAPS J. 14, 749– 758 (2012).
· 7 Meibohm, B. & Derendorf, H. Basic concepts of pharmacokinetic/pharmacodynamic (PD/PD) modeling. Int. J. Pharmacol. Ther. 35, 401– 413 (1997).
· 8 Csajka, C. & Verotta, D. Pharmacokinetic-pharmacodynamic modeling: history and perspectives. J. Pharmacokin. Pharmacodyn. 33, 227– 229 (2006).
· 9 Byon, W. et al. Establishing best practices and guidance in population modeling: an experience with an internal population pharmacokinetic analysis guidance. CPT Pharmacometrics Syst. Pharmacol. 2, e51 (2013).
· 10Guideline on Missing Data in Confirmatory Trials, EMA (2011). <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/09/WC50 0096793.pdf>
· 11 O’Neill, R.T. & Temple, R. The prevention and treatment of missing data in clinical trials: an FDA perspective on the importance of dealing with it. Clin. Pharmacol. Ther. 91, 550– 554 (2012).
· 12 The Panel on Handling Missing Data in Clinical Trials; National Research Council. The Prevention and Treatment of Missing Data in Clinical Trials (National Academies Press, Washington, DC, 2010). <http://www.nap.edu/catalog.php?record id=12955#orgs>
· 13 Holford, N. A time to event tutorial for pharmacometricians. CPT Pharmacometrics Syst. Pharmacol. 2, e43 (2013).
· 14 Hu, C. Exposure–response modeling of clinical end points using latent variable indirect response models. CPT Pharmacometrics Syst. Pharmacol. 3, e117 (2014).
· 15 Björnsson, M.A., Friberg, L.E. & Simonsson, U.S. Performance of nonlinear mixed effects models in the presence of informative dropout. AAPS J. 17, 245– 255 (2015).
· 16 Hu, C. & Sale, M.E. A joint model for nonlinear longitudinal data with informative dropout. J. Pharmacokinet. Pharmacodyn. 30, 83– 103 (2003).
· 17 Yang, J. et al. The combination of exposure-response and case-control analyses in regulatory decision making. J. Clin. Pharmacol. 53, 160– 166 (2012)
· 18 Goyal, N. & Gomeni, R. Exposure-response modeling of anti-depressant treatments: the confounding role of placebo effect. J. Pharmacokinet. Pharmacodyn. 40, 389– 399 (2013).
· 19 Hu, C., Wasfi, Y., Zhuang, Y. & Zhou, H. Information contributed by meta-analysis in exposure-response modeling: application to phase 2 dose selection of guselkumab in patients with moderate-to-severe psoriasis. J. Pharmacokinet. Pharmacodyn. 41, 239– 250 (2014).
· 20 Bouazza, N. et al. Concentration-response model of lopinavir/ritonavir in HIV-1-infected pediatric patients. Pediatr. Infect. Dis. J. 33, e213– e218 (2014).
· 21 Pouw, M.F. et al. Key findings towards optimising adalimumab treatment: the concentration-effect curve. Ann. Rheum. Dis. 74, 513– 518 (2015).
· 22 Wang, Y. et al. PK in late phase trials. Appl. Clin. Trials 21, (2012).
· 23 Karlsson, K.E., Vong, C., Bergstrand, M., Jonsson, E.N. & Karlsson M.O. Comparison of analysis methods for proof-of-concept trials. CPT Pharmacometrics Syst. Pharmacol. 2, e23 (2013).
· 24 Bhattaram, V., Siddiqui, O., Kapcala, L.P. & Gobburu, J.V.S. Endpoints and analysis to discern disease-modifying drug effects in early Parkinson’s disease. AAPS J. 11, 456– 464 (2009).
· 25 Lee, J.Y. Impact of pharmacometric analyses on new drug approval and labeling decisions: a review of 198 submissions between 2000 and 2008. Clin. Pharmacokinet. 50, 627– 635 (2011).
· 26 Overgaard, R.V. Exposure-response—does it have more to offer MBDD and how to get us there. American Conference of Pharmacometrics, Las Vegas (2014).
· 27Guidance for Industry, End-of-Phase 2A Meetings. Center for Drug Evaluation and Research, FDA (2009). <http://www.fda.gov/downloads/Drugs/…/Guidances/ucm079690.pdf>
· 28The Clinical Pharmacology and Biopharmaceutics (CPB) Review Template: The Question-Based Review (QBR). Center for Drug Evaluation and Research, FDA (2004). http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/StaffPoliciesandProcedures/ucm073007.pdf
· 29Good Review Practice: Clinical Review of Investigational New Drug Applications. Office of New Drugs in the Center for Drug Evaluation and Research, FDA (2013). http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/UCM377108.pdf
· 30Guidance for Industry, Exposure-Response Relationships—Study Design, Data Analysis, and Regulatory Applications. Center for Drug Evaluation and Research, FDA (2003). http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072109.pdf
· 31Concept paper on extrapolation of efficacy and safety in medicine development. EMA (2013). <http://www.ema.europa.eu/docs/en_GB/document_library/Other/2013/04/WC500142359.pdf http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/04/WC500142358.pdf>
· 32 Milligan, P. et al. Good practices in model informed drug discovery and development (MID3): practice, application, documentation and reporting. Poster presented at the American Conference on Pharmacometrics, Las Vegas, 2014.