文献:2006英国TGN1412事件:一期临床试验专家科学小组最终报告
付永超 / 2026-07-16
原文链接: https://webarchive.nationalarchives.gov.uk/ukgwa/20130105143109mp_/http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_073165.pdf 原文版权: Expert Scientific Group on phase one clinical trials: final report, © Crown copyright, licensed under Open Government Licence v3.0 **原文协议:**https://www.nationalarchives.gov.uk/doc/open-government-licence/version/3/
**译者注:**2006年发生在英国的一起TGN1412药物相关的首次人体试验事件是一个知名的事件,阅读调查该事件的报告原始资料可以丰富对首次人体试验的视角,译者因此翻译了此文件。翻译时主要通过必应翻译功能翻译,译者额外校对了部分词汇的中文翻译比如"pharmacodynamics"在本文应翻译为"药物效应动力学",而不是"药效学",以及比如"首次人体(FIH)",“最低预期生物效应水平(MABEL)",“药理学活性剂量(PAD)",“未观察到不良效应的水平(NOAEL)",“药物实体(agent)",“北威克公园医院(Northwick Park Hospital)“等。
一期临床试验专家科学小组
最终报告
2006年11月30日
由 TSO(文具办公室)出版,可通过以下方式获取:
在线:www.tsoshop.co.uk
邮寄、电话、传真及电子邮件
TSO
PO Box 29, Norwich, NR3 1GN
电话订单/一般咨询:0870 600 5522
传真订单:0870 600 5533
电子邮件:customer.services@tso.co.uk
文字电话:0870 240 3701
TSO 实体店
伦敦 123 Kingsway, WC2B 6PQ 020 7242 6393 传真 020 7242 6394
贝尔法斯特 16 Arthur Street, BT1 4GD 028 9023 8451 传真 028 9023 5401
爱丁堡 71 Lothian Road, EH3 9AZ 0870 606 5566 传真 0870 606 5588
TSO@Blackwell 及其他认证代理商
© 2006 王室版权 版权所有。
如需复制,请书面申请至版权部,英国皇家文具办公室,
St Clements House, 2-16 Colegate, 诺里奇 NR3 1BQ。首次出版 2006
ISBN-10 0 11 703722 2
ISBN-13 978 0 11 703722 9
在英国印刷,供文具办公室使用 5476378 C4 12/06
原始目录
- 执行摘要 页码 1
- 引言 13
- 截至2006年7月TGN1412事件背景 18
由ESG秘书处准备
A TGN1412临床前开发
B TGN1412向临床开发的过渡 24
C 北威克公园医院一期临床试验及不良事件 30
D 英国药品和医疗产品管理局(MHRA)GCP、GMP及GLP调查 37
- 《新英格兰医学杂志》发表的论文 44
- NIBSC特殊细胞因子研究及后续研究 56
- 利益相关者咨询总结 68
- 预测从临床前到临床的风险 75
- 风险降低与风险管理 83
- 建议范围及总结 97
- 附录
10.1 ESG会议、文件及会议记录 A 1 - 216
10.2 受邀利益相关者及咨询名单 B 1 - 3
10.3 利益相关者会议记录及展示 C 1 - 70
10.4 利益相关者书面提交
10.5 对中期报告的评论 D E 1 - 83 1 - 131
10.6 受邀观察员名单 F 1
10.7 观察员提交 G 1 - 2
10.8 英国一期临床试验历史 H 1 - 2
10.9 专家科学组成员 I 1 - 2
10.10 秘书处及专业支持人员 J 1
10.11 实验室分析结果表 K 1 - 3
- 术语表 L 1 – 7
简体中文翻译版详细目录
1. 执行摘要
背景
专家科学小组(ESG, The Expert Scientific Group)是在2006年3月TGN412"首次人体(first-in-man)“临床试验中发生非常严重的不良反应后成立的。这次试验是在伦敦"北威克公园医院(Northwick Park Hospital)“的一个私人临床研究单元进行的。TGN1412是一种单克隆抗体,正在开发为治疗白血病和自身免疫性疾病(如类风湿性关节炎)的药物。
在临床试验中,六名健康男性志愿者在静脉注射TGN1412后不久就遭遇了"严重的全身不良反应(severe systemic adverse)"。所有六名"志愿者(volunteers )“都出现了"细胞因子释放综合症(cytokine release syndrome)“并伴随"多器官衰竭(multi-organ failure)",需要密集治疗和护理,这些由北威克公园医院的重症治疗单元(ITU, Intensive Therapy Unit)提供。
以往的首次人体临床试验安全性记录都非常好,据我们所知,TGN1412试验中所有"受试者(recipients)“都经历如此严重且相似的不良反应,这是前所未有的。
在北威克公园医院发生这些事件后,健康大臣成立了ESG,其任务范围如下:
职权范围
- 考虑从临床前阶段向首次人体试验一期研究过渡时可能需要的事项,以及这些试验的设计,特别涉及:
x 具有新型作用机制的生物分子;
x 具有高度物种特异性作用的新药;
x 针对免疫系统靶点的新药。
- 向卫生大臣提供报告形式的建议,以便对此类试验的未来批准做出指导,中期报告需在三个月内提交。
ESG 被要求在编写报告前征求主要利益相关者的意见。
利益相关者提出了几个不在我们职责范围内的关注点。这些包括知情同意的流程和信息的清晰度、临床研究者与临床试验受试者在试验前和试验期间的沟通、保险覆盖情况、研究伦理委员会的作用,以及对经历不良反应的试验受试者的临床随访。虽然这些不在我们的职权范围内,但我们认为这些更广泛的关注点非常重要,并建议应将其作为高优先级来处理。
ESG的目标
ESG的目标是回顾可以从TGN1412试验中学到的经验,并提出建议,以提高未来首次人体试验中涉及我们参考条款所描述类型药物的安全性。
我们被要求在三个月内提交一份中期报告,并在制定建议时征询利益相关者的意见。在我们2006年7月26日发布中期报告后,我们又收到了来自利益相关者的38份书面和口头意见,包括四名试验志愿者及其代表、Northwick Park的医生、患者代表、个人、国内外公共机构、生物技术和制药行业以及合同研究组织。ESG已考虑这些书面意见和口头陈述,并在制定最终建议时予以参考。
在我们中期报告发布时,仍在等待由国家生物标准与控制研究所(NIBSC)进行的独立实验室测试的部分结果,以澄清TGN1412试验中出现的毒性。这些结果于2006年11月收到,并被仔细研究。新的实验结果可能为临床试验中TGN1412不良反应相关的科学问题提供潜在答案,同时解释为何在动物的临床前测试中未发现类似反应。这些结果已作为汇总部分被纳入我们的报告。
解决问题的方法
对更好、更安全药物的需求是显而易见的,同样显而易见的是,人类第一次"接触(exposure)“一种新药总会有一定风险,即使这个风险极小。我们的目标是对我们职权范围内的药物类型的首次人体试验,在优化首次人体试验安全性的同时不扼杀创新或增加开发有用新药的不必要障碍的。
ESG已召开了10次会议,举办了5次利益相关方会议,并考虑了利益相关方提交的52份书面材料和25份口头材料。
我们审查了TGN1412的临床前开发过程、MHRA调查结果以及试验中给药剂量导致未预测到的严重毒性的可能原因。我们的结论是,TGN1412所进行的临床前研究未能预测安全的人体使用剂量,尽管满足了现行监管要求。
给予人类志愿者的TGN1412剂量引发了细胞因子快速大量释放。细胞因子是传递免疫细胞与组织细胞信号的小型蛋白分子。细胞因子具有强大的生物活性,其产生通常受高度控制。细胞因子快速大量释放会引起发热、低血压和器官衰竭,这种现象被称为“细胞因子释放综合征(cytokine release syndrome)”或“细胞因子风暴(cytokine storm)”。这种情况发生在试验志愿者身上,但在用于计算"人体首次暴露(first human exposure)“的剂量的食蟹猴动物模型中并未发生。在比人类志愿者剂量高500倍的剂量下,食蟹猴也没有出现细胞因子释放综合征。
我们继续识别了与新药首次人体试验风险评估和风险管理相关的因素,这些因素在我们的职权范围内有说明。我们使用“药物实体(agent)”一词来描述正开发作为人用潜在药物的"化学或生物化合物(chemical or biological compound)"。
建议的适用范围
适用于什么类型的临床试验?
这些建议旨在适用于“首次人体”临床试验,而不是一般的一期临床试验(后者可能包括对已有人体安全记录药物的试验)。在首次人体暴露于高风险药物且剂量可能引起药理学作用时,需要特别谨慎。
然而,当将可能存在高风险的药物用于全新的特定人群时(无论是健康志愿者还是患者),也应该采取额外的谨慎措施。制定首次人体试验指南时,应考虑将已制定的概念应用于从一种人群过渡到另一种人群的情况。
ICH E8 指南《临床试验一般考虑(CPMP/ICH/291/95)(http://www.emea.europa.eu/pdfs/human/ich/029195en.pdf)》提供了关于临床试验的一般指导,其中第3部分包含了对一期试验的国际公认描述,以及从药物研发到上市各阶段的良好概述。
适用于什么类型的药物实体?
我们的权限涵盖三类药物,这些药物在首次人体"暴露(exposures)“时可能对志愿者带来更高的伤害风险,或者在临床前开发中风险可能更难评估。具体类别如下:
x 具有新作用机制的生物分子;
x 高度特异于某些物种的新药物实体;
x 以免疫系统为靶点的新药物实体。
我们希望我们的建议适用于这三类药物中的任何一种,除非经过仔细评估药物的性质、靶分子的生理作用以及预期使用者的情况后,确认首次人体暴露的风险较低。
我们并不建议所有属于上述三类的药物实体在首次人体暴露时一定会带来高风险,但在评估风险被认为较低时,应该进行彻底的风险评估,并提供清晰的科学依据。
例如,常规疫苗虽然旨在刺激免疫反应,但可能并不会带来高风险;或者类似于人体已有安全使用记录的新药物实体,且针对已知靶点,其药理作用可以有把握地预测,也可能不会带来高风险。
我们在本报告中讨论了在首次人体暴露新药物实体时应提高警惕的一些因素。包括:
x任何可能对重要身体系统造成严重生理干扰的因子;
x激动或刺激作用;
x新的药物实体和新颖的作用机制,且没有先前经验;
x特异性导致动物模型中的临床前风险评估困难或不可能;
x药物实体的"效力(potency)",例如与天然配体的比较;
x多功能的药物实体,例如二价抗体、FcR结合域;
x细胞相关靶点;
x靶点绕过常规控制机制;
x免疫系统靶点;
x靶点在具有体内大规模生物扩增潜力的系统中。
在首次人体试验之前,应该始终进行彻底的风险评估。风险评估应在试验文件中清楚说明,并由监管机构全面审查。如果存在重大疑虑,应始终假定风险较高。(见报告第7节)
建议
在临床试验中,受试者的安全、权利和福祉,无论是患者还是健康志愿者,都必须始终是首要考虑因素。我们提出了22条建议,我们认为这些建议将提高未来涉及首次人体暴露于潜在高风险药物的临床试验中志愿者的安全性,这些药物按照我们的职权范围分类,并在报告的第8节中进行了讨论。
这些建议包括:
x 临床前和早期临床阶段的开发;
x 临床试验申请的准备和审查过程,以及为监管机构和申办方提供的早期咨询机会;
x 确定和管理人体起始剂量;
x 首次人体试验的临床环境;
x 培养技能和培训以满足未来需求。
重点是分享与安全相关的信息、首次剂量的计算和管理,以及在评估首次人体暴露于我们职责范围内所述类型的新药试验申请时,获得独立专家意见的监管途径,这类试验具有较高的潜在风险。
每条建议的理由以及我们的意图说明在第7节“从临床前到临床转化的风险预测”和第8节“风险降低与风险管理”中都有概述。建议应结合这些章节来考虑。
我们的建议是针对英国当局和英国首例人体试验的申办方提出的,但我们认为重要的是应在欧盟及国际层面寻求共识,以确保全球"临床试验参与者(clinical trial participants)“获得同等保护。
致谢
我们特别感谢四位参与试验的志愿者,他们访问了ESG,并向我们提供了宝贵的个人经历分享。
我们还要感谢所有利益相关者,他们在向ESG提交的书面和口头意见中提供了许多深思熟虑且建设性的建议。
我们也感谢秘书处和技术支持团队,他们在准备背景文件、安排会议、记录会议纪要以及与国内外利益相关者沟通方面的辛勤工作,我们非常感激。
建议摘要
临床前和早期临床开发
- 关于新药临床前开发的策略,以及用于收集与首次人体试验安全性相关信息的实验方法的决定,必须基于科学,由具有"适当培训(appropriate training)“的个人逐案做出并"提供理由(justified)"。
新药的临床前开发是由国际公认的指南来规范的,特别是《ICH S6 生物技术衍生产品的临床前安全性评价》和《ICH M3 (R1) 药物开展人体临床试验的非临床安全性研究》(见下方进一步阅读)。
监管机构应考虑这些指南对于高风险药物,尤其是首次用于人体的药物的适用性。在当前背景下,应提出制定更具体指南的建议。这最好通过先在欧盟层面发布初步指南,然后以此为平台向国际层面提出建议来实现。
- 对于高风险药物实体和基于创新技术的先进药物的人体首次试验,其监管流程应该定期审查。
随着生物学、生物化学、信息学和生物技术的不断进步,我们可以预期会发现许多新的治疗靶点,并开发出满足未被满足临床需求的创新药物。药品监管必须以科学为基础,其监管流程也要跟上科学和技术的发展,并适应这些将带来未来药物的科学与技术。
- 药物开发者、研究资助机构和监管部门应加快收集与人体暴露安全相关的未发表临床前研究信息。第一步应聚焦于那些显示使用高风险药物或药物组合可能对人体造成危险的临床前研究报告。这将为欧盟及国际级别的监管部门之间的信息共享提供平台,例如以机密数据库的形式。强烈鼓励"研究者(investigators )“将这些数据提交到数据库中。
为了安全起见,我们认为最终目标应是一个开放访问的数据库,这一可行性需要进一步探讨。然而,为了避免延迟分享关键信息的风险,本应不公开的研究报告最初可以存储在仅全球监管人员可访问的安全数据库中,并需定期审查延迟开放的理由。
- 监管机构应考虑加快欧盟及全球监管机构之间一期临床试验安全性信息的共享。这当然应包括首次进入人体使用高风险药物的经验。应纳入具有负面安全性结果的试验。这个数据库可能会扩大,以包括目前可能不被视为高风险的产品,或在开发后期进行的试验,这些纳入的试验数据对首次使用类似产品提出了强烈警告。
在欧盟,这种信息收集和共享可以基于现有临床试验数据库EudraCT(自2004年以来的首次人体试验)和EudraVigilance数据库(疑似意外严重不良反应(SUSARs)的模型(见下文)。2004年前首次人体试验的相关信息可自愿提交。这将确保国家监管机构能够获取相关的安全信息。
这需要与制药行业对话,确定哪些研究报告应发布以及何时发布,但由于行业已承诺公开已上市药物的临床试验结果,并鼓励申办方发布可能存在重要安全隐患的开发失败结果,这应是可行的。监管机构应探讨开放访问这些数据的可行性。
SUSARs是疑似意外严重不良反应。它们之所以意外,是因为它们的性质或严重程度与适用的产品信息不符(例如,未经批准的研究产品的研究者手册或已经批准产品的产品特性总结),因为它们是与产品存在因果关系,且反应严重,如下所述
指令2001/20/EC第2(o)条对严重不良反应的定义:“任何不当的医疗事件或影响,在任意剂量下导致死亡、危及生命、需要住院或延长现有住院、导致持续或显著的残疾或丧失能力,或先天异常或先天缺陷。”
对于未列入本清单的其他重要医疗结果,在决定是否需要报告时,应进行专业的医学和科学判断。
临床试验申请的准备和审查过程,以及监管者和申办方的早期咨询机会
- 强烈建议开发者在提交申请前的早期阶段与监管机构进行更多沟通,特别是针对高风险试剂,以确保有足够时间适当考虑任何安全问题,同时不会对产品开发造成不必要的延误。也应考虑增加监管机构与研究伦理委员会之间的沟通方式。
对于属于我们职责范围内高风险类别的新药物实体首次人体临床试验,建议并强烈推荐申办方与监管机构进行"提交前会议(pre-submission meetings)",以便双方识别潜在问题。如开发者选择不与监管机构进行提交前讨论,应要求他们在拟提交高风险药物首次人体临床试验申请前六周通知监管机构。这将给予监管机构充分时间考虑是否需要外部专家意见,并确定合适的专家(见下一条建议)。提前通知可以减少延长评审过程的可能性。
- 对于高风险药物试验申请的评估,根据药物的性质、创新程度、预期药理靶点以及目标接受者,监管机构应该能够获得来自独立专业专家的额外意见,这些专家在其研究领域有丰富的知识。
- 人用药品委员会的专家咨询小组(EAG),或类似机构,可以承担这一角色,其核心成员由适当的专家组成,并能够根据需要吸收额外的专业知识。
- 在临床试验评估的时间安排上,对于一些异常复杂的特殊情况,应考虑引入一定的灵活性。
我们强调“异常复杂的特殊情况”;并无意在新药开发的监管过程中引入不必要的延误。《欧洲临床试验指令》(第6条第7款)(指令2001/20/EC,以下简称)已经允许对基因治疗、体细胞治疗以及含有转基因生物的产品的评估时间进行延长。这可能成为其他将新科学引入治疗的新型创新药物的参考模式。
确定并管理人体的起始剂量
- 对于那些其主要药理作用在动物模型中无法证明其拟议治疗效果的新药物,应给予特别关注。任何此类试验的剂量选择依据都应包括对拟议作用机制的明确理由,以及该物质在预期临床用途中的安全性和有效性的说明。
- 在剂量计算时,应采取比仅依赖动物研究中的“未观察到效应水平(NOEL, No Observable Effect Level)”或“未观察到不良效应水平(NOAEL, No Observable Adverse Effect Level)”更广泛的方法。起始剂量的计算应利用所有相关信息。需要考虑的因素包括药物的新颖性、生物活性及作用机制、药物的物种特异性程度、人类和动物细胞生物效应的剂量-反应曲线、体内动物实验的剂量-反应数据、药物代谢动力学和药物效应动力学建模、靶点占有率与浓度的计算以及人体体内靶点或靶细胞的暴露量的计算。
“最低预期生物效应水平(MABEL, minimum anticipated biological effect level)”方法是实现这一目标的一个很好的模型。(参见BIA/ABPI报告及利益相关者提交。)
- 如果不同的方法给出的人体安全剂量估计不同,首次人体试验的起始剂量应该取最低值,并在计算实际起始剂量时引入"安全冗余量(margin of safety)"。
- 当临床前信息出于任何原因,可能无法很好地指导人体体内反应时,首次人体试验的起始剂量应以谨慎为原则进行计算。之后的剂量增加也应小心进行,因为起始剂量可能特别低,而且剂量-反应曲线可能很陡峭。
- 在首次人体试验中,应该仔细考虑首个剂量的给药途径和速度,并密切监测是否出现"不良反应或过度反应(adverse or exaggerated response)"。
例如,对于首次静脉注射高风险药物的人体暴露,几小时的缓慢输注可能比几分钟的缓慢推注更合适。这可以让医生监控不良反应,并在临床上有必要时停止输注。
- 试验设计,包括受试者人数、起始剂量的决定以及剂量递增方案,应该根据具体情况来制定,并且在科学和统计上都有合理依据,同时考虑所有相关信息。
(参见英国皇家统计学会的相关意见)
- 在首次人体试验中,新药物应按顺序给不同受试者使用,并在每次给药之间留有适当的观察时间。受试者之间的观察间隔应根据药物的性质、靶点和受体特征,以及药物的潜在药代动力学、毒理动力学来预估可能出现的不良反应。
不同受试者之间以及同一受试者的重复给药之间都应留有合适的间隔,并且间隔时间都应基于现有证据加以合理说明。
- 在剂量递增的过程中,受试者之间连续给药时,也应进行类似的监测周期(如上所述)。
- 是否在健康志愿者或患者志愿者中进行首次人体试验,应经过仔细考虑并充分论证,同时考虑所有与受试者安全性相关的因素以及可能获得的科学信息价值。
一般来说,在新药的首次人体试验中,患者没有预期的直接获益。因此,风险与收益的评估通常不是决定是否应在健康志愿者或患者志愿者中进行此类试验的主要因素。
在癌症药物领域中可能存在例外情况,因为经历试验药物有效反应的患者通常能够继续使用该药。在这种情况下,风险与收益的平衡可能会成为决定由患者作为临床试验对象的因素,尤其是未对现有疗法有反应的患者,以及当试验药物具有可预测的细胞毒性(实际上可能是其预期的药理作用)时。
然而,最重要的因素应始终是志愿者的权利、安全和福祉,无论是患者还是健康个体,以及从临床试验中可以获得的信息价值。
首次人体试验的临床环境
- 负责首次人体试验受试者护理的主要研究者应始终具备适当的资格,并确保自己对试验药物、其作用靶点和作用机制有足够了解,从而能够做出知情的临床判断。
应大力鼓励为进行首次人体临床试验的主要研究者建立国家级职业认证体系。
- 在首次人体研究中,如果可预见会出现某些类型的严重不良反应,应提前考虑治疗策略。这应包括在有特定解毒剂可用时确保其可用性,并制定明确的支持性治疗计划,包括预先安排的重症监护(ITU)设施应急可用性。
- 对于高风险药物的首次人体试验,应始终在适当的临床环境中进行,由具备相应培训和专业知识的工作人员监督,并且应能立即使用用于处理和稳定急性紧急情况的设施,同时应事先安排好合理距离内的重症监护室(ICU)预案。
首次人体试验中应始终保证足够的工作人员配置,并在志愿者过夜时提供充足的24小时保障。
所有开展此类试验的临床地点都应有应急情况的标准操作程序,工作人员应通过定期演练保持执行这些程序的专业能力。试验参与者应始终被清楚告知在临床试验期间或之后出现不良反应症状时应该怎么做。
发展专业技能
- 应该扩大在临床试验的规划和执行中获得“实践”经验的机会。例如,研究生培训项目可以设有派遣到商业机构的交流期,或者在 NHS 和大学的专业中心进行培训的阶段(见下一条建议)。
英国医学科学院 2005 年发布的《更安全的药物》报告指出,需要培训新一代医生掌握药物安全性和有效性评估所需的技能。在最近 ABPI 进行的一项研究报告《维持制药和生物制药行业技能储备》中也发现了类似需求。
这需要高等教育资助机构和院校、NHS 以及行业之间的合作。鉴于主要利益相关方一致认同需要通过培养新一代临床药理学家、毒理学家及相关专业人员来填补这一技能缺口,这一问题应该可以得到解决,我们鼓励这样做,以确保未来首例人体临床试验的安全。
- 应探索建立针对高风险药物和先进药物产品的一期临床试验专科中心的可行性。
应鼓励开发国家级临床中心检查和认证系统,专门针对开展高风险药物人体首次试验的中心。认证应向所有符合规定标准的中心开放,包括公立和私立部门。
延伸阅读(Further reading)
‘Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Monoclonal Antibody TGN1412’
G. Suntharalingam, M. Perry, S. Ward, S. Brett, A. Castello-Cortes, M. Brunner, and N. Panoskaltsis.
N Engl J Med. 2006, 355:1018-1028.
The European Clinical Trials Directive 2001/20/EC (http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol- 1/dir_2001_20/dir_2001_20_en.pdf)
EudraLex Volume 10 –Clinical Trials (http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev10.htm)
European Union Guidelines - http://www.emea.europa.eu/htms/human/humanguidelines/background.htm
ICH E8 - General Considerations for Clinical Trials (CPMP/ICH/291/95) (http://www.emea.europa.eu/pdfs/human/ich/029195en.pdf)
ICH S6 Pre-clinical Safety Evaluation of Biotechnology-Derived Products (CPMP/ICH/302/95)
(http://www.emea.europa.eu/pdfs/human/ich/030295en.pdf)
ICH M3 (R1) Non-Clinical Safety Studies for the Conduct of Human Clinical Trials for Pharmaceuticals (CPMP/ICH/286/95). (http://www.emea.europa.eu/pdfs/human/ich/028695en.pdf).
ICH International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) http://www.ich.org/cache/compo/276-254-1.html
Academy of Medical Sciences 2005 report “Safer Medicines” (http://www.acmedsci.ac.uk/p102.html).
“Sustaining the skills pipeline in the pharmaceutical and biopharmaceutical industries”, ABPI. (http://www.abpi.org.uk/Details.asp?ProductID=285).
延伸阅读(翻译了文献标题)
‘在抗CD28单克隆抗体TGN1412一期临床试验中的细胞因子风暴’ G. Suntharalingam, M. Perry, S. Ward, S. Brett, A. Castello-Cortes, M. Brunner, 和 N. Panoskaltsis. 《新英格兰医学杂志》. 2006, 355:1018-1028.
欧洲临床试验指令 2001/20/EC (http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol-1/dir_2001_20/dir_2001_20_en.pdf)
EudraLex 第10卷 – 临床试验 (http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev10.htm)
欧盟指南 - http://www.emea.europa.eu/htms/human/humanguidelines/background.htm
ICH E8 - 临床试验的一般考虑 (CPMP/ICH/291/95) (http://www.emea.europa.eu/pdfs/human/ich/029195en.pdf)
ICH S6 生物技术衍生产品的临床前安全评估 (CPMP/ICH/302/95) (http://www.emea.europa.eu/pdfs/human/ich/030295en.pdf)
ICH M3 (R1) 药物开展人体临床试验的非临床安全研究 (CPMP/ICH/286/95) (http://www.emea.europa.eu/pdfs/human/ich/028695en.pdf)
ICH 国际人用药品注册技术要求协调会议 (ICH) http://www.ich.org/cache/compo/276-254-1.html
医学科学院 2005 报告 “更安全的药物” (http://www.acmedsci.ac.uk/p102.html)
“保持制药和生物制药行业技能储备”,ABPI. (http://www.abpi.org.uk/Details.asp?ProductID=285)
2. 介绍
北威克帕克医院一期临床试验的不良事件
2006年3月13日,六名健康男性志愿者在Parexel(一家合同研究机构)组织的一项一期首次人体临床试验中接受了TeGenero的TGN1412药物,另外两名志愿者服用了安慰剂。该Parexel单位位于伦敦北威克帕克医院的租用场地。TGN1412是一种单克隆抗体,正在开发为治疗B细胞白血病和自身免疫性疾病的新药。在接受TGN1412几小时内,所有六名志愿者因出现非常严重的全身炎症反应被送入北威克帕克医院重症监护室,病情发展为多器官衰竭。
专家科学小组(ESG)的成立
ESG 由卫生国务大臣于2006年4月成立,此前在TGN1412临床试验期间,Northwick Park医院发生了悲惨事件。
ESG 的职权范围
在TGN1412临床试验之后,卫生国务大臣任命ESG,赋予其以下任务:
- 考虑从临床前阶段向首次人体试验一期研究过渡时可能需要的事项,以及这些试验的设计,特别涉及:
x 具有新型作用机制的生物分子;
x 具有高度物种特异性作用的新药;
x 针对免疫系统靶点的新药。
- 向卫生大臣提供报告形式的建议,以便对此类试验的未来批准做出指导,中期报告需在三个月内提交。
ESG 被要求在剔除建议之前征求主要利益相关者的意见。
ESG由17位来自科学、临床医学、药理学、毒理学、免疫学、生物伦理学、临床试验、药物开发和药物监管背景的成员组成。此外,还有两名普通成员,总共19人。ESG还接待了来自日本、美国和欧盟药品监管机构的观察员,以及卫生部、贸易与工业部和基因治疗咨询委员会的观察员。
ESG 的目标、工作方式和会议安排
使用职权范围中描述的药物实体类型,目标是优化首次人体试验安全性,而不会对有用新药的开发造成不必要的障碍。
在对 TGN1412 试验中的事件进行评估后,ESG 与国家生物标准与控制研究所(NIBSC)进行了联络,以独立完成 TeGenero 在 TGN1412 早期临床开发中进行的一些关键实验,并对 TGN1412 的生物特性进行进一步研究。
考虑到 TGN1412 临床试验的结果以及已知的 TGN1412 单克隆抗体信息,ESG 思考了在一期试验中可能普遍重要的先验风险识别因素。
ESG 识别并讨论了 TGN14 的一些可能带来风险的特性,并指出这些特性可能可以推广到其他首次人体临床试验的药物实体,例如:
x 不同物种间作用的特异性给临床前开发带来挑战;
x 在生物放大级联中的靶点上具有激动作用;
x 免疫系统靶点;
x 能激活信号通路的细胞表面靶点;
x 多功能分子,例如可能同时激活带 FcR 的细胞和主要靶细胞的抗体;
x 效力:
- 与天然配体相比,对靶受体具有更高亲和力、占有率或信号效应(例如“超激动抗体”);
- 通过正常(如抗体)或改变的药代动力学(如聚乙二醇化)延长在靶点的暴露时间;
- 多价性,可以实现靶分子的交联。
这些以及与药物实体本身性质和其靶点生理功能相关的其他因素,都进行了详细考虑,试图理解哪些内容可以概括出来,以便识别我们权限范围内描述的类别中更高风险的药物实体。
随后,我们详细探讨了与新药从临床前到临床开发过渡过程中不同方面相关的风险降低和风险管理问题,重点关注首次人体暴露。讨论的问题示例包括:
临床前研究
x 如何"验证(validate)“动物模型的"预测价值(predictive value)"?
x 有没有方法可以优化体外实验对人类细胞/组织的"预测价值(predictive value)"?
x 开发关于动物模型对新药物实体或药物类别反应的经验共享数据库的可行性和价值如何?这种数据库是否可以由国际监管机构、制药和生物技术公司、学术研究人员、科研伦理委员会共同贡献并访问?
x 开发计算机模拟;它现在有多大用处,或者未来可能在什么时候有用?
临床
x 剂量安全裕度充足;NOAEL或动物模型中的NOEL是否是新型药物的有效起点?考虑到所有相关信息,人类的安全裕度是否足够大?
x 高风险药物的顺序给药在首次人体试验中是否必要?第二批及后续受药者需要等待多久才能获得给药?
x 新药剂的给药途径和速率有多重要,尤其是具有新机制的药物?
x 如何选择第一个受试者?如何确保知情同意?
x 何时以及如何递增剂量?
x 即使在一期试验中,健康志愿者和患者的风险/收益比存在差异吗?首次人体试验是否应包括健康志愿者或患者志愿者?这是具体情况问题吗?
x 一期试验的临床环境应该是什么样的?
x 足够的员工、专业知识和培训的重要性?
x 是否应该有更多培训机会来获得相关技能?
x 高风险药物的首次人体试验是否需要在医院现场进行?
x 复苏和重症监护中心设施的随时可用性?
x 预期出现重大不良反应及其类型?
x 针对潜在ADR且可识别风险的特定疗法的可用性?
x是否向主要研究者披露了足够的数据,以允许临床判断?
x 是否应该为高风险或非常创新药物的首次人体试验设立专科单位?
x之前临床试验经验的信息共享是否足够?
x 临床试验安全性数据库可以实现开放获取吗?
监管
x 对于高风险的“首次人体”临床试验申请,是否应该由独立专家进行审查;例如由CHM的专家咨询小组审查和/或在需要时增加高度专业化的专家?
x 是否可以用实用的方式来定义高风险药物实体?
x 在现行时间范围内(在申请处理期间或提交申请之前)应该如何执行?
x 时间尺度应该延长,还是时钟可以被停止?
会议和关键日期(2006年)
| 5月23日 | ESG 会议。 |
|---|---|
| 来自秘书处的关于 TGN1412 开发及临床试验中不良反应的背景资料。 | |
| MHRA 调查报告 | |
| 北威克公园 ICU 医生的临床报告 | |
| 5月31日 | ESG 会议。 |
| 不良反应的机制 | |
| 风险因素识别 | |
| 高风险药物识别 | |
| 6月2日 | ESG 会议。 |
| 风险降低策略 | |
| 6月14日 | ESG 会议。 |
| 风险降低策略(续) | |
| 利益相关者意见 | |
| 6月15日 | 与利益相关者的小组会议 |
| 6月16日 | 与利益相关者的小组会议 |
| 6月19日 | 与利益相关者的小组会议 |
| 6月20日 | 与利益相关者的小组会议 |
| 6月27日 | ESG 会议。 |
| 对 TGN1412 试验利益相关者意见的评估 | |
| 7月11日 | 与利益相关者的 ESG 会议。 |
| 利益相关者意见,临时建议 | |
| 7月20日 | 作为咨询文件发布的中期报告。 |
中期报告作为咨询文件发布后,于2006年9月、10月和11月邀请了利益相关者进一步的陈述和书面提交。还举行了以下会议:
| 9月12日 | 与利益相关者的 ESG 会议。 |
|---|---|
| 10月24日 | 与利益相关者的 ESG 会议。 |
| 11月13日 | 与利益相关者的 ESG 会议。 |
| 11月14日 | 与利益相关者的小组会议,包括四名临床试验志愿者及其代表。 |
| 11月15日 | ESG 会议。 |
| NIBSC 结果 | |
| 讨论利益相关者意见 | |
| 最终建议 | |
| 11月30日 | 提交部长的最终报告 |
在提交最终报告和建议之前,ESG 详细考虑了所有利益相关者的意见。
3. 截至2006年7月TGN1412事件的背景。由ESG秘书处准备
A TGN1412的临床前开发
CD28在免疫系统中的作用
CD28是一种共刺激受体,表达在CD4 T淋巴细胞(T细胞)表面以及大部分CD8 T细胞上。它能够有效地与T细胞抗原受体(TCR)的信号结合,共同对静息T细胞进行共刺激。CD28受体促进TH1和TH2细胞的发展。TH1和TH2细胞是辅助T细胞,帮助白细胞活化反应。TH1细胞在宿主防御中促进“细胞免疫”,并参与像移植排斥这样的反应。TH2细胞则通过帮助B淋巴细胞(B细胞)增殖和分泌抗体来促进“体液免疫”。
在自然状态下,CD28信号通路的激活需要TCR被抗原触发的同时,CD28也被其生理膜结合配体B7-1(CD80)或B7-2(CD86)激活。在体外,这一过程可以通过使用针对TCR和CD28的特异性抗体组合来模拟。激动性抗CD28的单克隆抗体TGN1412可以绕过触发TCR信号的要求,激活人类T细胞,而不考虑它们TCR的特异性。正因为这个特性,它被称为“超激动剂(superagonist’)”。
研究发现,早期的TCR信号对于由激动型抗CD28的抗体介导的T细胞增殖并非必需,这一点在人类和大鼠T细胞上都得到了验证——见图1。
进一步研究表明,像TGN1412这样的激动型抗CD28单克隆抗体只结合在CD28免疫球蛋白样胞外结构域侧面暴露的C’’D环上,而传统的共刺激抗体则识别靠近天然配体结合位点的表位,这种特异性与抗CD28抗体的激动活性密切相关。最近,利用X射线晶体学分析也再次确认了C’’D环对激动型抗CD28单克隆抗体结合的重要作用。
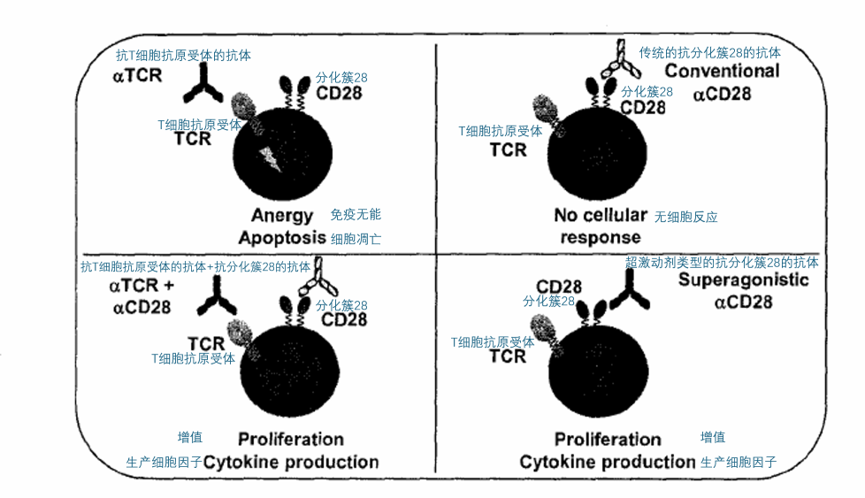
图1:在没有TCR刺激的情况下人类T细胞的激活。 TGN1412绕过了触发TCR信号的需求,在没有TCR刺激的情况下激活人类T细胞。在T细胞中,仅TCR触发会导致免疫无能和凋亡。普通的抗CD28的抗体无法诱导T细胞的反应。通过抗TCR的抗体和抗CD28的抗体进行同时触发,可以在体外导致增殖和促炎细胞因子的分泌,但在体内则不行。相比之下,TGN1412在体外可以显著促使T细胞增殖,并在体内能也能够良好地扩增T细胞。
TGN1412 人源化单克隆抗体
TGN1412 是一种激动型抗 CD28 单克隆抗体,开发用于各种疾病的治疗,这些疾病中 T 细胞参与慢性炎症或血液系统恶性肿瘤如白血病的发病机制。该抗体是一种重组人源化单克隆抗体,能够特异性结合存在于 T 细胞上的 CD28。TGN1412 是通过将单克隆小鼠抗人 CD28 抗体重链和轻链可变区序列中的互补决定区 (CDRs) 转入人源重链和轻链可变区框架中进行基因工程改造的。随后,人源化的可变区分别与编码 IgG4 重链的人基因和编码轻链的人基因重组。人源恒定区和可变区框架结构预计可以降低免疫原性,并在人体免疫系统内实现抗体效应功能的最佳化。
TGN1412 分子由两条约 24 kDa(214 个氨基酸)的轻链和两条约 51 kDa(447 个氨基酸)的重链组成。该蛋白在 CHO 细胞中表达,分子量约为 148 kDa。TGN1412 “药物产品(drug product)“是用于输注的缓冲等渗无防腐剂浓缩液。在"临床材料(clinical material)“中,“原料药(drug substance)“的浓度为10 mg/ml,装入 40 ml 小瓶。容器为 50 ml 注射瓶。
开发TGN1412用于血液系统恶性肿瘤的理由是基于它能够重建一个崩溃的T细胞群(例如在B-CLL中)。在使用来自多种B-CLL患者的原始血液样本进行的离体实验中,已经证明TGN1412能够诱导多克隆T细胞的扩增和活化。TGN1412无需TCR触发就能激活T细胞,不论其TCR特异性如何。这种新的T细胞激活方式被称为“激动型(agonistic)”或“超激动型(superagonistic)”。在引用的文献中,这两个术语是同义使用的。
此外,研究间接显示TGN1412可以改善BCLL细胞抗原呈递的不足。TGN1412假定的免疫调节(抗炎)特性与它激活和扩增调节性T淋巴细胞以及诱导抗炎性细胞因子的功能有关。激动型抗CD28治疗在多种自身免疫疾病的动物模型中被证明有效,包括类风湿关节炎动物模型、大鼠实验性自身免疫性神经炎(EAN)以及大鼠实验性自身免疫性脑脊髓炎(EAE)。
毒理学报告
使用动物和人类细胞进行体外开发
在多种检测系统中(包括流式细胞术和Biacore分析)显示了TGN1412对人类CD28的特异性。TGN1412不会与密切相关的受体细胞毒性T淋巴细胞抗原-4(CTLA-4, Cytotoxic T-Lymphocyte- Antigen-4)和诱导共刺激分子(ICOS, Inducible Costimulator)发生交叉反应。
对激动型抗CD28抗体的拓扑学要求已经通过使用嵌合CD28分子进行表位绘图进行了详细研究。研究表明,特异性针对大鼠或人CD28的激动型抗体仅与CD28免疫球蛋白样结构的外侧暴露C’’D环结合,而常规的共刺激抗体则识别接近天然CD80/CD86配体结合位点的表位。此外,经过改造表达人的C’’D环序列的小鼠CD28分子,在被激动型抗人CD28抗体交联时,即使不进行TCR连接也能激活T细胞,这表明激动功能与C’’D环存在因果关系。最近,通过X射线晶体学分析也进一步确认了C’’D环在激动型抗CD28单克隆抗体结合中的关键作用。因此,基于共享的特异性-功能关系,激动型抗大鼠CD28单克隆抗体JJ316以及抗人CD28激动剂5.11.A1、TGN1112和TGN1412被认为是正宗的同源物。
研究还使用TGN1412和TGN1112进行了实验,以演示它们与来源于啮齿动物和非人灵长类动物(如恒河猴Macaca mulatta (rhesus monkey)、食蟹猴Macaca fascicularis (cynomolgus monkey) 和绒猴Callithrix jacchus (marmoset monkey))T细胞上表达的CD28的交叉反应,以支持选择合适物种进行安全性和毒理学研究的合理性。
TGN1412 对人类、食蟹猴和恒河猴的T细胞都有反应,其反应模式是CD28特有的。而对绒猴的T细胞,尚未观察到结合反应。
此外,还对不同物种CD28的C’’D环进行了氨基酸序列同源性分析。人类和食蟹猴的C’’D环序列完全一致,与恒河猴CD28仅一个氨基酸不同,而绒猴C’’D环在6个氨基酸中有2个不同。啮齿动物的C’’D环与人类CD28的C’’D环同源性很低。
TGN1412 激活 T 细胞的能力是在体外增殖实验中建立的,这些实验使用了外周血单核细胞(PBMC)、纯化的 T 淋巴细胞以及高度纯化的 T 细胞亚群,如 CD4+和 CD8+T 细胞、初始 CD4+CD45RA+和记忆 CD4+CD45R0+细胞,或者来自健康供体的常规 CD4+CD25- 以及调节性 CD4+CD25 高 T 细胞。将 PBMC 与可溶性 TGN1412 共孵育会导致多克隆 T 细胞增殖和 T 细胞特异性细胞因子的分泌。不同血液供体的 TGN1412 诱导增殖程度存在差异,而常规的、共刺激的人特异性抗 CD28 抗体通常无法引发显著的细胞增殖。因此,TGN1412 被认为在通过 CD28 传递有丝分裂信号而无需 TCR 共结合方面具有独特性。
动物模型中的体内开发
一项对恒河猴进行的纵向研究,用于评估TGN1412和IgG1变体TGN1112的耐受性及T细胞激活效果。TGN1112能够在体内激活和扩增T细胞。重要的是,对恒河猴施用具有激动作用的抗CD28单克隆抗体,并未导致系统性细胞因子(IL-5、IL-6、IL-10、IFNγ)血清浓度发生显著变化,也没有出现长期(最长至第155天)副作用。
观察发现,TGN1412在恒河猴中的药理活性明显弱于TGN1112。这可以通过两种抗体类型在亲和力和/或Fc受体结合特性上的差异来解释。
TGN1412的T细胞激活特性是在食蟹猴中进行的研究中确定的。流式细胞术结果显示,CD4和CD8 T细胞显著扩增,且在输注后大约第15天达到高峰。短暂的T细胞扩增伴随着细胞激活,表现为CD69和CD25的升高。
TGN1412 的药代动力学和毒代动力学
由于蛋白质降解途径众所周知,所以没有对 TGN1412 进行常规的吸收、分布、代谢和排泄研究。
在比较佐剂性关节炎大鼠模型和恒河猴中的结果时,发现替代性促激动性抗 CD28 抗体(JJ316 和 TGN1112)的动力学存在差异。这些差异在 JJ316 和 TGN1112 诱导的体内 T 细胞扩增动力学中至少部分得到了解释。这种差异是预料中的,因为它反映了啮齿动物和非人灵长类动物之间的普遍差异。
因此,在食蟹猴中测得的 TGN1412 毒代动力学特性,被认为最能预测人类的药代动力学。
在食蟹猴的重复剂量毒性研究中,毒代动力学评估显示,TGN1412的血清浓度随时间变化的曲线总体上与药物静脉(iv)注射一致。在首次注射5 mg·kg^-1后,TGN1412的终末消除半衰期被估计约为8天,这与抗体等大分子生物分子相对缓慢的清除速度一致。
在一只动物中,观察到TGN1412血清浓度相对较低,这归因于抗-TGN1412抗体的存在。在TGN1412报告的毒代动力学参数中,并未发现与性别相关的一致性差异。
随着剂量从5 mg·kg^-1增加到50 mg·kg^-1,TGN1412的系统暴露量增加了大约20倍。此外,还有证据表明,随着剂量增加,TGN1412的平均终末半衰期也有所延长。
由于TGN1412并不直接作用于恶性组织,因此药物效应动力学效应预计不会仅仅与血浆/血清中活性物质的浓度相关,而是取决于其对外周血和淋巴组织中T细胞子集的影响。在这方面,需要注意的是,尽管在连续四周的TGN1412注射后观察到了四次血清峰值,但T细胞扩增只出现了一次(大约在第15天)。T细胞不重复扩增的原因可能是由于CD28抗原的可调性、TGN1412靶结构的可获得性、T细胞的功能性非应答性和/或T细胞扩增受到稳态机制的限制。
非临床安全性研究
由于TGN1412在CD28外域上的表位仅限于人类和非人灵长类动物,食蟹猴和恒河猴被认为是评估TGN1412对人体潜在毒性的最相关物种。
非临床毒理学计划包括重复剂量毒性研究、局部耐受性研究以及与人类和食蟹猴组织交叉反应性的免疫组织化学研究。这些研究均按良好实验室规范(GLP)进行。静脉给药途径被选择,以符合临床试验中的预期应用。
这些研究结果显示,TGN1412在食蟹猴中每周连续四周给药至50 mg·kg-1时耐受良好。在这些研究中,没有观察到TGN1412相关的毒性、过敏反应或全身免疫系统异常的迹象。对主要生理系统(心血管系统、呼吸系统和中枢神经系统)也没有观察到不良影响。
因此,50 mg·kg-1被认为是”未观察到不良效应水平(NOAEL, no-observed-adverse-effect level)”。
未进行生殖和发育毒性研究。然而,在食蟹猴28天毒性研究中,对生殖道组织进行了组织病理学检查,没有观察到与处理相关的变化。
由于TGN1412的生物特性,标准的基因毒性测试被认为不合适,因此未进行。
在接受治疗的食蟹猴注射部位观察到的局部反应被认为与TGN1412治疗无关,而是与给药操作有关。在兔子的局部耐受性研究中,TGN1412通过静脉、外周静脉或动脉内给药均能良好耐受,且未产生临床显著的刺激反应。
TGN1412的免疫毒性在标准毒理学研究和非GLP药理学研究中进行了评估。对于补充研究,使用了特异性针对大鼠CD28的激动性抗体(JJ316)或TGN1412的IgG1变体(TGN1112)。给非人灵长类动物注射TGN1412或TGN1112后,在给药后第13至17天之间,CD4+和CD8+ T细胞数量出现短暂升高,这是TGN1412预期的药物效应动力学效应。个别动物在TGN1412治疗下,血清IL-2、IL-5和IL-6水平中度升高,但未出现首次给药的细胞因子释放综合征(CRS)的临床症状。此外,在用任何剂量的激动性抗CD28单克隆抗体治疗的动物中,没有发现过敏反应、自身免疫疾病诱发或意外的系统性免疫抑制的证据。
在组织交叉反应性研究中,淋巴细胞染色的分布与淋巴组织中T细胞预期的分布一致(靶抗原特异性)。在人类和食蟹猴供体的中枢神经组织中,还观察到额外的特异性染色,认为可能代表星形胶质细胞染色。然而,中枢神经组织的交叉反应性并未与食蟹猴的中枢神经相关的不良临床症状或毒理学发现相关联。在食蟹猴供体的子宫颈和人类胎盘的细胞滋养层细胞中记录到细胞质内染色。此类细胞质内染色被认为临床上不重要,因为细胞质抗原的暴露似乎是组织切片导致的。28天毒理学研究中食蟹猴生殖道没有报告与治疗相关的组织病理学发现,这进一步支持了这一假设。
References
- Riley J. and June C. The CD28 family: a T cell rheostat for therapeutic control of T cell activation. Blood, 2005; 105: 13 to 21.
- Dennehy K.M., Kerstan A., Bischof A., Park J. H., Na S. Y., and Hunig, T. Mitogenic signals through CD28 activate the protein kinase Ctheta-NF-kappaB pathway in primary peripheral T cells. Int. Immunol., 2003; 15: 655 to 663.
- Luhder F., Huang Y., Dennehy K., Guntermann C., et al. Topological requirements and signaling properties of T cell-activating, anti-CD28 antibody superagonists. J. Exp. Med., 2003; 197: 955 to 966.
- Evans E., Esnouf R., Manso-Sancho R., Gilbert R., et al. Crystal structure of a soluble CD28-Fab complex. Nature Immunol., 2005; 6: 271 to 279.
- Schmidt J., Elflein K., Stienekemeier M., Rodriguez-Palmero M., et al. Treatment and prevention of experimental autoimmune neuritis with superagonistic CD28-specific monoclonal antibodies. J. Neuroirnmunol., 2003; 140: 143 to 152.
- Beyersdorf N., Gaupp S., Balbach K., Schmidt J., et al. Selective targeting of regulatory T cells with CD28 superagonists allows effective therapy of experimental autoimmune encephalomyelitis. J Exp. Med., 2005; 202: 445 to 455.
参考文献
- Riley J. 和 June C. CD28 家族:调控 T 细胞活化的 T 细胞调节器。Blood, 2005; 105: 13 到 21。
- Dennehy K.M., Kerstan A., Bischof A., Park J. H., Na S. Y., 和 Hunig, T. 通过 CD28 的有丝分裂信号在原代外周 T 细胞中激活蛋白激酶 Ctheta-NF-kappaB 通路。Int. Immunol., 2003; 15: 655 到 663。
- Luhder F., Huang Y., Dennehy K., Guntermann C., 等。T 细胞激活抗 CD28 抗体超级激动剂的拓扑要求和信号特性。J. Exp. Med., 2003; 197: 955 到 966。
- Evans E., Esnouf R., Manso-Sancho R., Gilbert R., 等。可溶性 CD28-Fab 复合物的晶体结构。Nature Immunol., 2005; 6: 271 到 279。
- Schmidt J., Elflein K., Stienekemeier M., Rodriguez-Palmero M., 等。用超级激动的 CD28 特异性单克隆抗体治疗和预防实验性自身免疫性神经炎。J. Neuroimmunol., 2003; 140: 143 到 152。
- Beyersdorf N., Gaupp S., Balbach K., Schmidt J., 等。使用 CD28 超级激动剂选择性地靶向调节性 T 细胞,实现实验性自身免疫性脑脊髓炎的有效治疗。J Exp. Med., 2005; 202: 445 到 455。
B TGN1412 向临床开发的转化
使用人类志愿者
基于拟议的 TGN1412 作用机制以及其在非人灵长类动物中已显示的临床前安全性,公司认为在免疫功能正常的人群中分析其免疫学安全性和药物效应动力学效应,为选择健康受试者作为 TGN1412 首次人体试验的目标群体提供了依据。
特别是,公司认为在健康受试者中进行的研究在患者研究之前,有助于获得有关 TGN1412 安全性和药理学的宝贵信息,这是合理的,因为:
i. 靶抗原 CD28 在健康受试者以及类风湿关节炎(RA)和B细胞慢性淋巴细胞白血病(B-CLL)患者中表达相当,这意味着安全性、药代动力学(PK)和药物效应动力学(PD)数据至少部分可以转移到 B-CLL 和/或 RA 患者身上。
ii. 在健康受试者中,TGN1412 的免疫安全性被认为最有可能没有问题,因为在多项前临床研究中,包括非人灵长类动物,健康动物没有观察到不良反应。但在已有免疫病理倾向的受试者中,免疫不良反应不能完全排除。
iii. 健康受试者的安全性和药理数据解释不会因为 T 细胞的预激活或功能障碍,也不会因为效应/记忆 T 细胞与调节性 T 细胞或造血系统其他组成部分的预先不平衡而受到影响。
iv. 如研究方案所述,健康受试者代表一个相对同质的人群,能够避免潜在混杂因素的影响,比如预先/同时用药和/或疾病活动性,从而更好地解释 TGN1412 的安全性和药理学。
剂量计算
由于TGN1412的特异性显然仅限于人类和非人灵长类T细胞上表达的CD28,因此在非人灵长类(食蟹猴和恒河猴)中进行的安全性和毒理学研究被认为是评估TGN1412对人类潜在毒性最相关的研究。为了确定与TGN1412作用机制相关的潜在不良反应以及意外毒性,研究人员在非人灵长类中开展了多项TGN1412的安全性和有效性研究,采用了单次给药和多次给药方案。这些研究结果显示,TGN1412在连续至少4周,每周高达50 mg·kg^-1的剂量下耐受性良好。
I期临床试验起始剂量的计算主要基于食蟹猴重复给药毒性研究中的未观察不良反应水平(NOAEL),以及美国食品药品监督管理局(FDA)草案指南《在成人健康志愿者中进行治疗性药物临床试验中估算安全起始剂量》所述程序。NOAEL被认为是50 mg·kg^-1。
根据FDA指南,对食蟹猴的NOAEL应用了3.1的全ometric校正因子,以计算“人体等效剂量”(HED),得出16 mg·kg^-1。当随后应用默认的安全系数10时,最大推荐起始剂量(MRSD)估计为1.6 mg·kg^-1。公司随后再增加了额外的安全裕度,提出起始剂量为0.1 mg·kg^-1。
拟议I期试验中的剂量范围被认为确保了最大患者安全,因为使用了保守的160的安全系数,同时也能研究TGN1412的基础PD/PK特性,这对于未来在患者人群中进行的临床试验至关重要。
使用“最小预期生物效应水平”(MABEL)方法
在恒河猴和食蟹猴中观察到了TGN1412及其替代物的药理活性,剂量在2.5到25 mg·kg^-1之间。从在健康和关节炎(佐剂性关节炎)大鼠中使用大鼠CD28特异性同源抗体JJ316进行的前期研究中,可以得出未观察效应水平(NOEL)<0.3 mg·kg^-1。最佳药理反应剂量范围在1到5 mg·kg^-1之间。因此,最小预期生物效应水平(MABEL)剂量可认为在0.3到1 mg·kg^-1之间。如果将MABEL认为是0.5 mg·kg^-1,并利用CPMP关于单次微剂量给药支持临床试验的非临床安全研究立场文件中概述的安全标准,则安全起始剂量将被计算为0.005 mg·kg^-1或5 µg·kg^-1。
两个产品在健康受试者中作为“首次人体试验”的起始剂量计算示例,用以与 TGN1412 进行比较。
产品 1
MABEL
在人类血浆中体外测定的最大抑制率约为 40%,浓度为 0.2 ug/mL。在 2 ug/mL或 20 ug/mL的浓度下,抑制程度没有变化。计算得出导致 20% 抑制的浓度为 0.01 ug/mL。使用血浆体积为 50 ml/kg,这相当于剂量为 0.5 ug/kg。从猴子的毒代动力学研究来看,5 ug/kg的静脉推注剂量会导致血浆峰值浓度为 189 ug/mL。0.5 ug/kg剂量的预期最大浓度是 0.0189 ug/mL。
起始剂量 0.5 ug/kg
人体等效剂量(HED)
基于最敏感物种的 NOAEL,通常将 HED 的1/10作为健康志愿者的最大安全起始剂量。如果 NOAEL 尚未确定,应额外增加最多10倍的安全系数。
计算方法如下:
猴子观察到的不良反应剂量:5 mg/kg
相应的人体等效剂量(HED):1.6 mg/kg
HED的1/10:0.16 mg/kg
额外的10倍安全系数:0.016 mg/kg
起始剂量:16 ug/kg
微剂量
计算出会产生药理作用的剂量是0.5mg/kg。微剂量的定义是低于计算出会产生药理作用剂量的1/100。也就是5 ug/kg。
起始剂量 5微克/公斤
产品 2
MABEL
该化合物在体外与其啮齿动物同源物相比,亲和力和效力没有差异。两种分子在啮齿动物疾病模型中都有效。
测试化合物的最低有效剂量为 0.3mg/kg。这对应于啮齿动物血浆中的浓度为 0.04mg/ml。使用血浆体积 50ml/kg,这折算成剂量为 2mg/kg。
起始剂量 2mg/kg
人体等效剂量(HED)
通常,基于最敏感物种的 NOAEL 的 HED的 1/10 被认为是健康志愿者的最大安全起始剂量。如果 NOAEL 尚未确定,还应增加最多 10 的额外安全系数。
计算如下:
大鼠和猴子的 NOAEL 都是 500 mg/kg
对应的人体等效剂量 (HED):大鼠 81 mg/kg
猴子 162 mg/kg
HED 的 1/10:8.1 mg/kg
起始剂量 8 mg/kg
微剂量
计算出会产生药理作用的剂量是5mg/kg。微剂量的定义是低于计算出会产生药理作用剂量的1/100。这就是50ug/kg。
起始剂量 50ug/kg
TGN1412 的受体占有率计算
受体占有率与局部浓度的例子
| 解离常数 | 1.88E-9 (mol/L) | TGN1412 的Kd |
|---|---|---|
| 分子量(道尔顿) | 150000 | TGN1412 的分子量 |
| 分布容积(毫升/千克) | 57 |
| 剂量 | 血浆浓度 | 受体占有率 |
|---|---|---|
| (mg/kg) | (M) | (%) |
| 0.0001 | 1.17E-11 | 0.006 |
| 0.001 | 1.17E-10 | 0.059 |
| 0.01 | 1.17E-09 | 0.384 |
| 0.1 | 1.17E-08 | 0.862 |
| 1 | 0.000000117 | 0.984 |
| 10 | 0.00000117 | 0.998 |
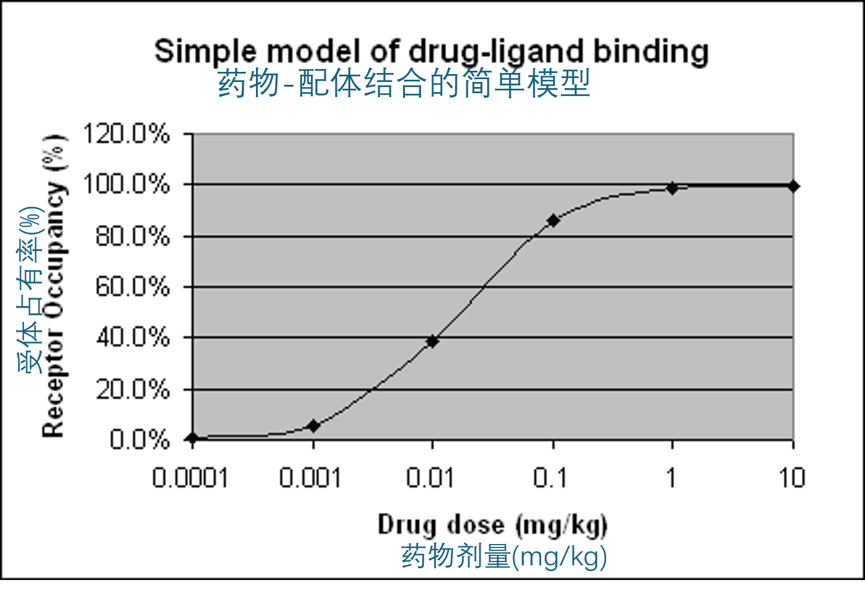
TGN1412的受体占用率,由ABPI/BIA工作组计算
TGN1412:一个例子
可以通过ABPI/BIA联合文件中概述的简单关系来估算单克隆抗体(mAb)与其靶点结合的量,其中:
mAb(A)与配体(B)处于平衡状态形成mAb-配体复合物(C)。结合亲和力(Kd)表示为 [A] [B]/[C],mAb通常选择高亲和力的,即它们更倾向于与靶配体结合。
在体内,mAb和靶配体的浓度会随时间变化。然而,作为初步估算,可以通过假设以下条件来估计循环血液中与靶结合的mAb的量(或百分比):
i) 靶配体(CD28受体)没有周转(清除)
ii) mAb不会因分布和清除而损失
iii) 结合反应瞬时达到平衡。
虽然这些假设并不能真实反映体内情况,但它们可以给出用药后最初几小时内可能与可用靶结合的mAb的大致估计。
剂量 TGN1412 0.1 mg/kg
体重 70 kg
分子量 TGN1412 150000
血容量 5L 血浆容量 2.5L
T淋巴细胞计数基线值(给药前)= 1.3 x 10^9 cells每L血液
CD28受体 150000每个T细胞 (Bryl et al 2001; 167 (6): 3231-3238)
Kd 1.88 nM (TeGenero, 公开资料)
给药后即刻的血浆中TGN1412总浓度 (A+C) 18.7 nM
基线时暴露于血浆的总配体 (CD28) 浓度 (B+C) 0.648 nM,假设 B+C = 1.3 x 10^9 x 150,000 (receptors/cell)/NA x 10^9【译者注:= 1.3 x 10^9 x 150,000/(6.02x 10^23) x 10^9 =0.324nM,该值于文中的值查了两倍,NA在此处为阿伏伽德罗常数6.02*10^23】
用药后即刻的药物-配体浓度 (C) 0.587 nM
TGN1412占据的CD28受体百分比 90.6%
显然,这个初始剂量预计会在给药后立即使CD28受体的占用率非常高,并可能达到最大的药理效果。事实上,即使很小剂量的单克隆抗体(低于临床推荐剂量)也可能达到最大的药理效果,尽管持续时间较短。药物效果的持续时间主要由单克隆抗体的剂量和靶配体的更新速率决定。对于像TGN1412这样的受体激动剂,这种受体占用水平应结合临床前药理研究中得到的结果,以及动物与人体之间的相对效力来考虑。这些信息尚未公开。
一些简化的一般原则
要精确估计初始配体结合,需要先估计靶标浓度。在某些情况下,这可能很难,例如当配体存在于组织中或体积不固定时(如滑液)。当配体浓度预计明显低于抗体时,即 A » B,对于单克隆抗体,配体的分数占有率(Ro)大致可以通过以下公式给出:
Ro = 1/(1 + Kd[nM] / (187[nM/mg/kg] x Dose[mg/kg])
单克隆抗体分布相对变化不大,这意味着187这个因子对于大分子蛋白(分子量超过约10万道尔顿)是通用的。作为一个参考,起始剂量用mg/kg表示的话,按Kd[nM]/200计算,初始配体占有率大约为50%。对于符合ESG参考条款的抗体,比如TGN1412,初始占有率越低越好。受体初始占有水平应由申请人在IMPD申请中说明。例如,用这种方法,0.001 mg/kg的TGN1412剂量预计受体占有率在5-10%之间。
References
- Estimating the Safe Starting Dose in Clinical Trials for Therapeutics in Adult Healthy Volunteers. U.S. Department of Health and Human Services Food and Drug Administration, Centre for Drug Evaluation and Research (CDER), Centre for Biologics Evaluation and Research (CBER), December 2002
- CPMP Position Paper on Non-Clinical Safety Studies to Support Clinical Trials with a Single Microdose. CPMP/SWP/2599/02. January 2003
参考文献
- 在成人健康志愿者中估算治疗性药物临床试验的安全起始剂量。美国卫生与公共服务部食品药品管理局,药物评价研究中心(CDER),生物制品评价研究中心(CBER),2002年12月
- 支持单一微剂量临床试验的非临床安全研究的CPMP立场文件。CPMP/SWP/2599/02,2003年1月
C 一期临床试验及北威克公园医院的不良事件
引言
TGN1412,一种CD28超级激动剂,原计划在位于北伦敦北威克公园医院租赁空间的Parexel临床药理研究单位(CPRU)进行“首次人体”试验。Parexel CPRU是负责TGN1412试验的合同研究机构。TGN1412或其类似物曾在临床前研究中测试过以下动物:大鼠(JJ316)、兔子、恒河猴和食蟹猴(TGN 1112和TGN 1412)。TGN 1112是TGN 1412的一种IgG1变体。
5.1 试验设计
拟议的试验是一个I期、单中心、双盲、随机、安慰剂对照、剂量递增的分组研究,旨在评估单次静脉注射TGN1412对健康志愿者的安全性、药代动力学、药物效应动力学和免疫原性。
主要目标是评估不同队列中TGN1412单次给药剂量递增的安全性和耐受性,并评估药代动力学。
次要目标包括评估TGN1412对淋巴细胞亚群、细胞因子谱以及单次TGN1412注射后抗TGN1412抗体产生的影响。
参与者计划在第1天入住CRPU,并留在单位至第3天。在输注开始后05、1、2、4、8和12小时测量生命体征。之后计划在指定间隔内进行进一步(随诊)记录,直到第43天。
拟议的剂量范围为静脉注射0.1、0.5、2.0和5.0 mg/kg(每kg体重)或安慰剂。
5.2 试验方案
纳入与排除标准(按方案定义)。
使用预先设定的一组标准来选择合适的志愿者;纳入标准:
▪ 18-40岁健康男性
▪ 体质指数18-28 kg/m²
▪ 根据病史、体格检查、心电图和实验室指标判断身体健康
▪ 筛查和第(-1)天C反应蛋白(CRP)正常,以排除炎症过程
▪ 能够提供知情同意
排除标准:
适用多种排除标准,其中较为显著的包括:
o 有任何感染或炎症(根据CRP)
o 淋巴结肿大
o 有重大系统性疾病史(心肺、神经等)
o 有多种药物过敏史或任何临床过敏性疾病
o 艾滋病、乙型或丙型肝炎
o 过去4个月内接受过研究药物或疫苗的受试者
o 本研究中曾接受过前一次剂量(TGN 1412)的受试者
o 酗酒或物质滥用,精神能力障碍,过去4天内进行剧烈运动,高血压,心率低等情况
在方案中提供的风险最小化措施
方案列出了以下潜在的不良事件,并计划了在发生此类情况时的一般和特定治疗方法;
| 事件 | 一般措施 | 具体措施 |
|---|---|---|
| 过敏反应 | 气道/循环支持/血管收缩剂 | 皮质类固醇 / β受体激动剂 / 抗组胺药 |
| 免疫原性/自身免疫 | 避免进一步暴露 | 如有需要,使用类固醇/抗组胺药。 |
| 一般皮肤反应 | 抗组胺药、止痛药 | 如有必要,使用皮质类固醇 |
| 细胞因子释放综合症 | 一般支持性措施 | 皮质类固醇及其他适当的临床措施。 |
| 严重不良事件 | 一般支持性措施 | 系统特异性措施。如果报告了多起严重不良反应,停止试验。 |
| 免疫抑制(大量诱导抗炎细胞因子和/或调节性T细胞) | 适当的对策 | 感染等情况使用抗生素。 |
停止受试者参加试验的原因:
受试者退出;
o撤回同意
o不可容忍的不良事件
o并发疾病或手术并发症
o违反方案入组研究或研究期间违反方案
o研究者对受试者最大医疗利益的意见
o新数据的可用性引发了对药物安全性的担忧
提前终止试验;
o不良事件的数量和严重程度证明了停药的合理性(研究者和申办者认为)
o新数据的出现引发了对药物安全性的担忧
5.3 不良事件:
报告来源于 SUSARs。
第一次筛选日期为 2006 年 2 月 22 日。首次给药日期为 2006 年 3 月 13 日。
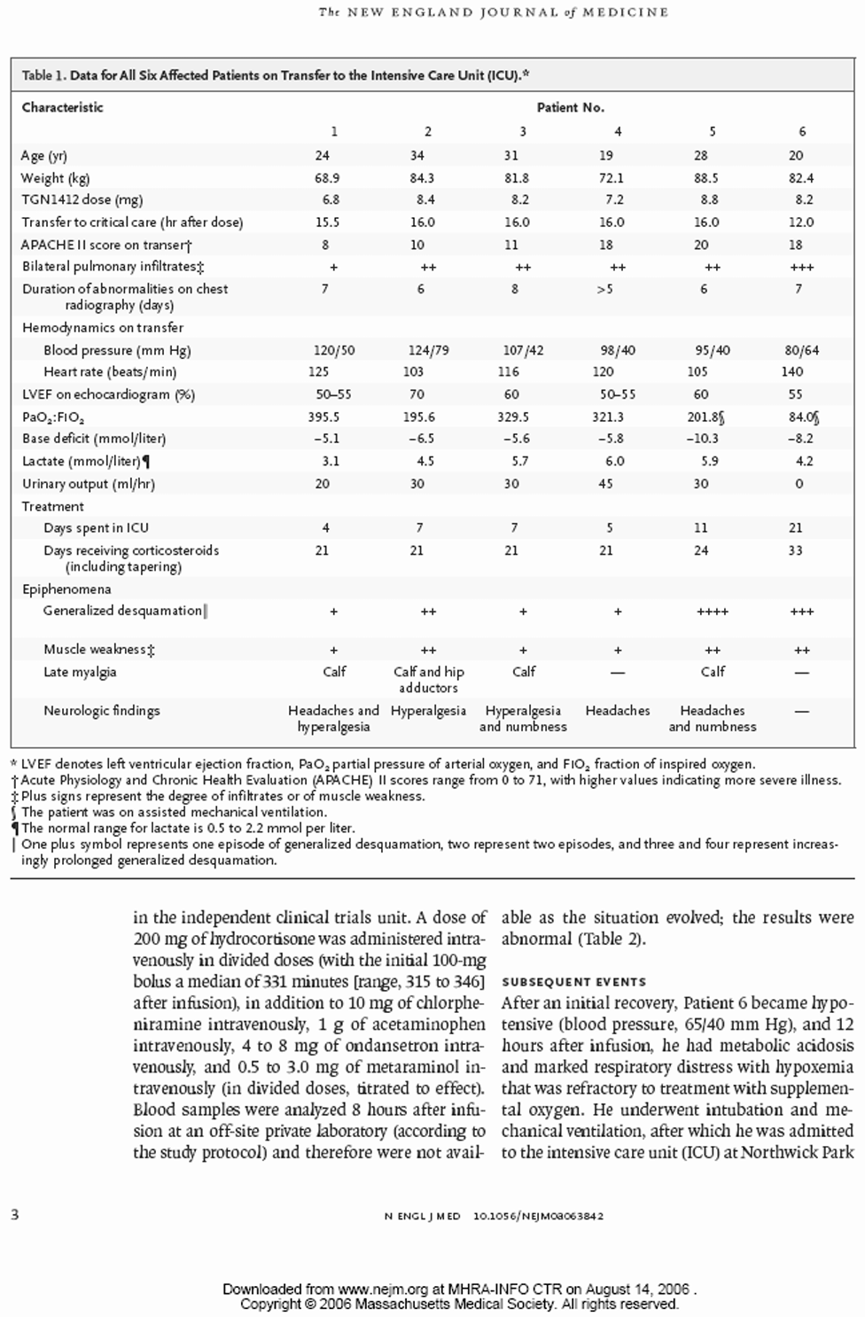
招募了 8 名年龄在 19-34 岁的受试者(平均 26 岁)。所有人在给药前的血液学和生化指标均正常。
给药前的参数
| Subjects → | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 尿素Urea (mmol/L) | 6.0 | 3.6 | 4.6 | 4.3 | 5.8 | 4.9 |
| 肌酐Creatinine (umol/L) | 89 | 82 | 79 | 74 | 81 | 78 |
| 中性粒细胞Neutrophils[x 10^9 /L] | 2.32 | 1.73 | 2.54 | 3.69 | 5.14 | 2.16 |
| 淋巴细胞Lymphocytes [x 10^9 /L] | 1.47 | 1.63 | 2.28 | 1.69 | 2.59 | 2.03 |
| C反应蛋白CRP (units) | <5 | <5 | <5 | <5 | <5 | <5 |
| 谷丙转氨酶ALT | 23 | 22 | 32 | 25 | 36 | 25 |
给药剂量和时间
所有受试者于2006年3月13日上午8点到9点之间按约10分钟间隔给药。六人接受了活性药物,两人接受了安慰剂。
| Subjects → | 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|---|
| 体重Body weight (Kg) | 84.3 | 68.9 | 88.5 | 82.4 | 72.1 | 81.8 | |
| 给药Dosing | |||||||
| TGN1412 | 8.4 mg | 6.8 mg | 8.8 mg | 8.24mg | 7.2 mg | 8.2 mg | |
| 给药时间Dosing time | 08:00 | 08:20 | 08:30 | 08:40 | 08:50 | 09:00 |
症状和发生时间
在使用研究药物(药物/安慰剂)后不久,受试者报告了多种症状。
x 在6名受试者中有5人报告出现头痛,时间为50-90分钟。
x 所有人都报告有腰部肌肉酸痛。
x 4名受试者报告在58-120分钟出现寒战。
x 体温升高>38°C的情况出现在2.5到6.5小时之间。
x 低血压出现在给药后3.5到4.6小时之间。
x 心动过速出现在2.5到4.6小时之间。
其他报告的症状包括恶心、呕吐、呼吸困难和肠道不适。一些受试者随后报告感到极度不适,并且无法按特定顺序回忆事件(报告为健忘)。
初步治疗
所有受试者都接受了静脉输液(总量 4 到 8 升;胶体 1.5 到 4.0 升,晶体液 1 到 4 升)。所有受试者还使用了血管收缩剂(美他扎明 1 毫克)和镇痛药(包括口服对乙酰氨基酚和可待因)。
所有受试者在症状发作后 4 到 6 小时内都服用了皮质类固醇(氢化可的松 100 毫克)。
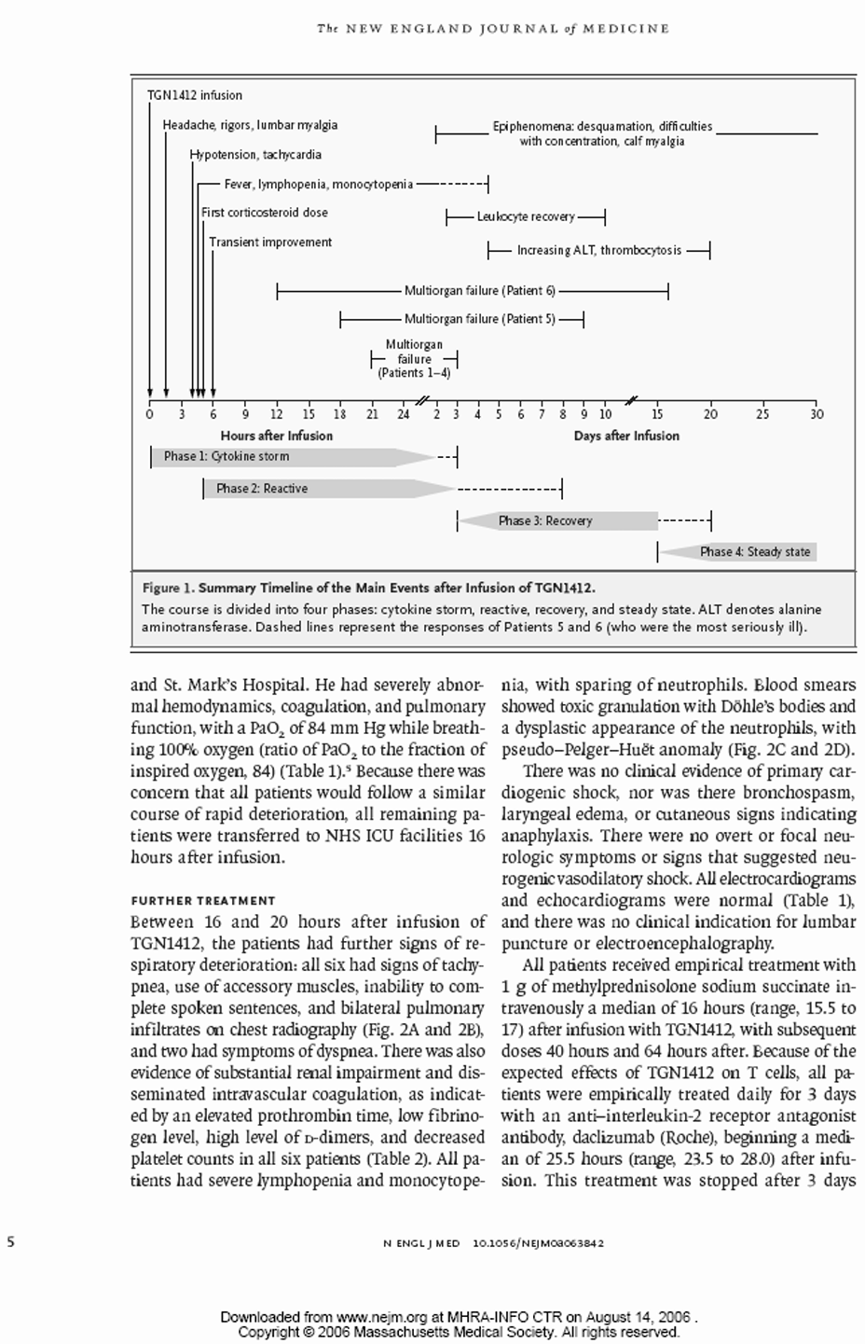
转入重症监护
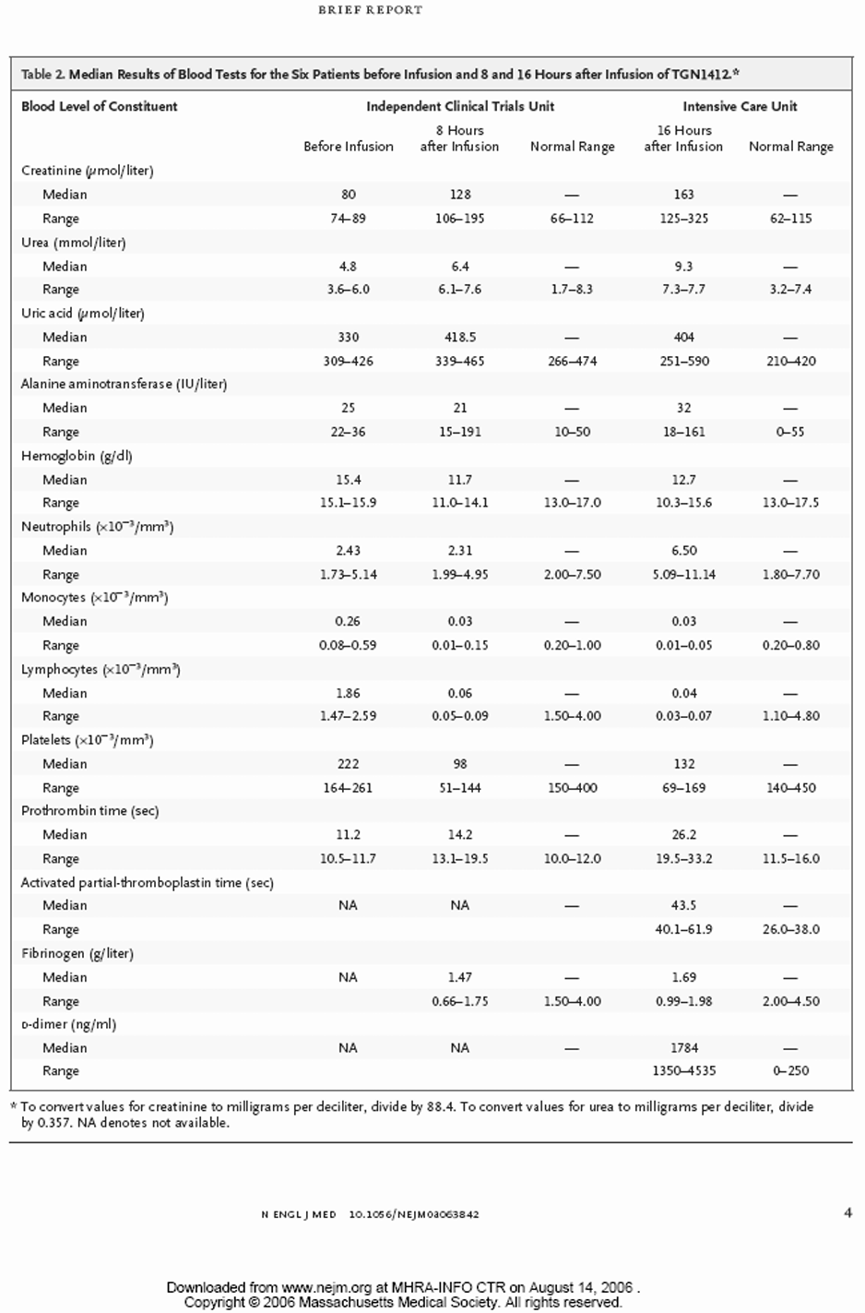
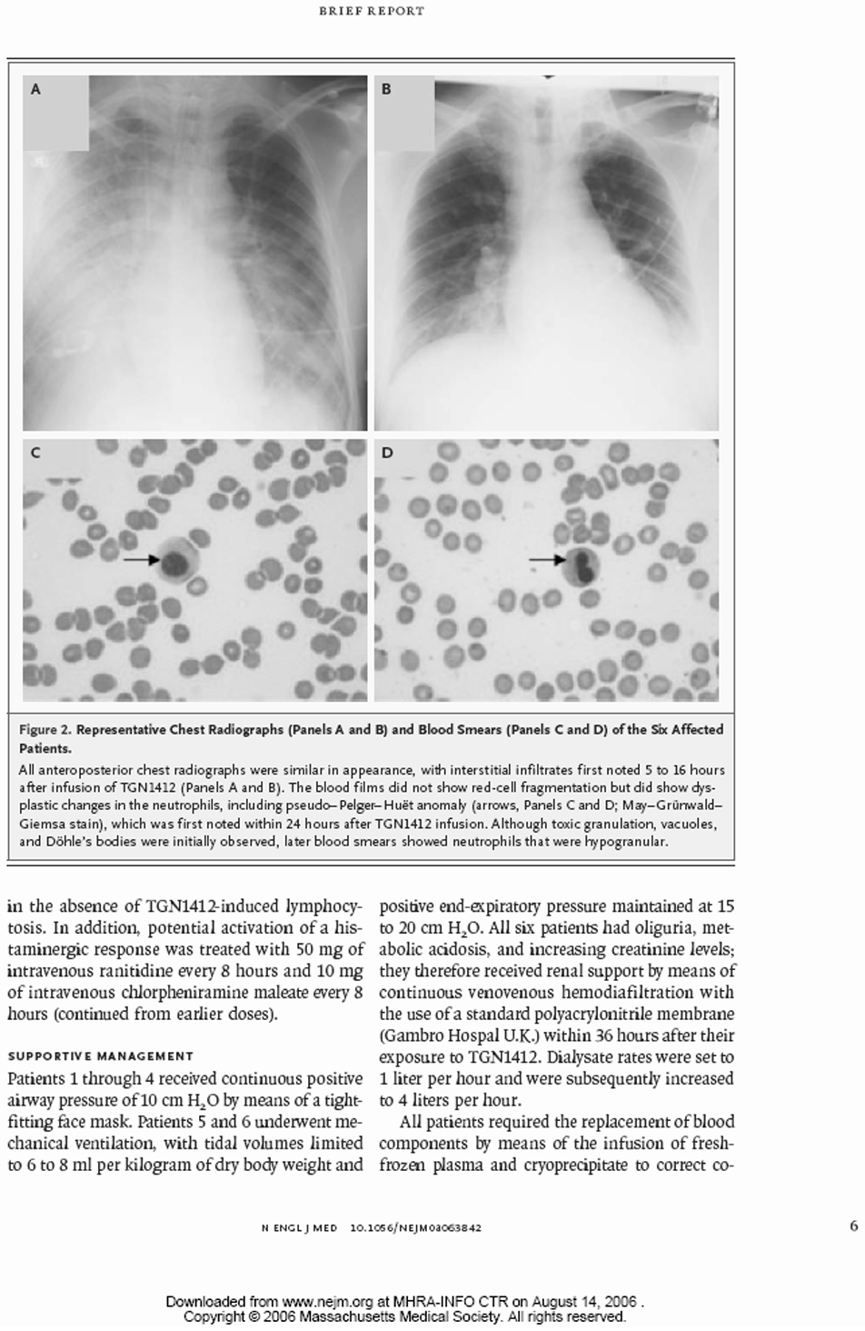
所有6名有症状的受试者在给药后12到16小时内因临床状态持续恶化,被转入Northwick Park医院的重症监护病房。在转入重症监护病房时,主要和常见的特征包括:6人中有5人出现呼吸困难和呼吸急促,6人中有4人出现呼吸衰竭,所有人双侧影像学肺部浸润,血尿素升高,明显碱缺乏,以及符合弥散性血管内凝血的特征(纤维蛋白降解产物升高,纤维蛋白原低,凝血酶原时间改变)。与给药前(基线1.47至2.59 x 10^9/L)相比,重症监护病房内普遍淋巴细胞减少(值在0.04至0.07 x 10^9/L之间),且这一状况持续了好几天。
重症监护中的治疗
x 呼吸支持:
所有受试者(患者)都需要辅助通气:在6名未插管的患者中,有4名接受了持续气道正压通气(CPAP),持续时间为4到82小时。有两名受试者在给药后12到18小时使用了间歇性正压通气(IPPV),这两名受试者都需要IPPV。
x 静脉-静脉滤过:
在重症监护病房(ITU)中,所有受试者都使用了持续静脉-静脉滤过(CVVHDF),使用时间不同,范围从44小时到158小时。两名受试者出现少尿或无尿。
x 类固醇/免疫抑制剂:
在首次剂量后,所有受试者都接受了第二剂氢化可的松(每剂100毫克,总计200毫克),随后又给所有受试者静脉注射了三剂甲泼尼龙(每剂1克),第一次在给药后16小时,随后在40小时和64小时分别给药。每位受试者还给了三剂达克珠单抗(Daclizumab,一种抑制T淋巴细胞增殖的免疫抑制单克隆抗体,剂量为1mg/kg)。上述治疗后,低剂量类固醇仍继续使用,持续21到33天不等。
其他临床事件
在两名受试者中观察到心房颤动和外周四肢缺血;
出现了全身性脱屑(3名受试者持续时间较长)、肌肉无力、肌肉疼痛和痛觉过敏。还注意到其他神经系统现象,包括头痛、注意力不集中、记忆力下降和肢端麻木。
并发症/其他事件
六名受试者中有三人在住院期间感染(克雷伯菌、念珠菌、金黄色葡萄球菌足部伤口感染),另外两名受试者发生病毒再激活(口鼻疱疹及其他部位疱疹性损伤),这些情况对相应治疗(抗生素和大剂量阿昔洛韦)有反应。
临床随访
在所有6名受试者中,发热(体温>38°C)的持续时间在14小时到36小时之间。在进入重症监护病房(ICU/重症治疗室)时,所有受试者都需要使用血浆容量扩充剂、血管收缩剂以及静-静脉血液滤过,持续时间为44到158小时。其中3名受试者的重症治疗室停留时间较长。
TGN引发的临床事件解读
在使用TGN1412后的事件与另一种药物OKT3抗体观察到的情况类似。OKT3在给人类及某些动物使用时,会引起细胞因子风暴,从淋巴细胞/单核细胞及其他细胞释放各种炎症性细胞因子。
接受TGN1412的6名受试者出现的事件也呈现类似的临床表现,包括全身症状(肌肉痛、头痛、低血压和心动过速)。这些症状随着前文描述的细胞因子和淋巴细胞亚群的变化而进一步发展。
细胞因子释放
细胞因子的测定是评估 TGN1412 反应方案的一部分。细胞因子结果是从不同实验室获得的。这里总结展示了一个私营机构的数据。Northwick Park 医院免疫学和血液学实验室的数据以非数字形式列在附录中。申办方公司(TeGenero)的数据目前还不可用。
检测的细胞因子包括:TNF-α、IFN-γ、IL-2、IL-4、IL-6 和 IL-10。
细胞因子表
| PREDOSE | 1 HOUR | 4 HOURS | DAY 2 | DAY 3 11:50 | DAY 11:50 | DAY 4 | DAY 5 | DAY 6 am | |
|---|---|---|---|---|---|---|---|---|---|
| Mean TNFalpha; | <2.8 | 1943 | > 5000 | 836 | 107 | 136 | <2.8 | <2.8 | 3 |
| Mean IFNγ | <7.1 | 99 | > 5000 | 4730 | 1366 | 270 | 89 | 43 | 27 |
| MeanIL-10 | <2.8 | 76 | 2158 | 1771 | 272 | 69 | 19 | 10 | 8 |
| Mean-IL-6 | < 3.0 | 29 | 1330 | 1204 | 96 | 475 | 466 | 95 | 43 |
| Mean-IL4 | <2.6 | 9 | 1205 | 13 | 24 | 3 | 3 | 3 | 3 |
| Mean-IL2 | 4.7 | 57 | 3317 | 137 | 14 | 9 | 4 | 3 | 4 |
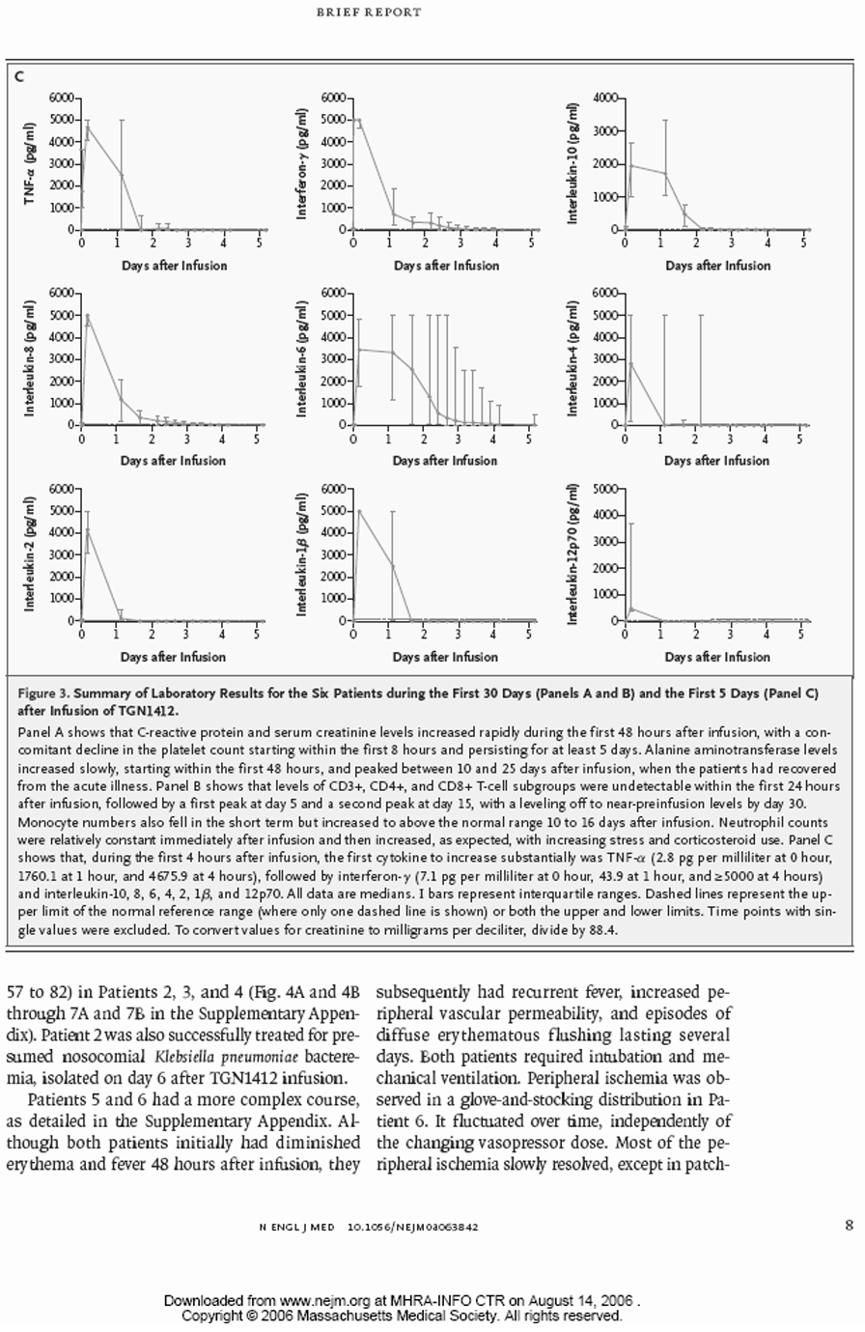
表格只显示四舍五入到最接近整数的平均值。这仅用于提供细胞因子值变化的示意表示。符号(<)和(>)表示值低于或高于检测范围,因此不是精确数值。
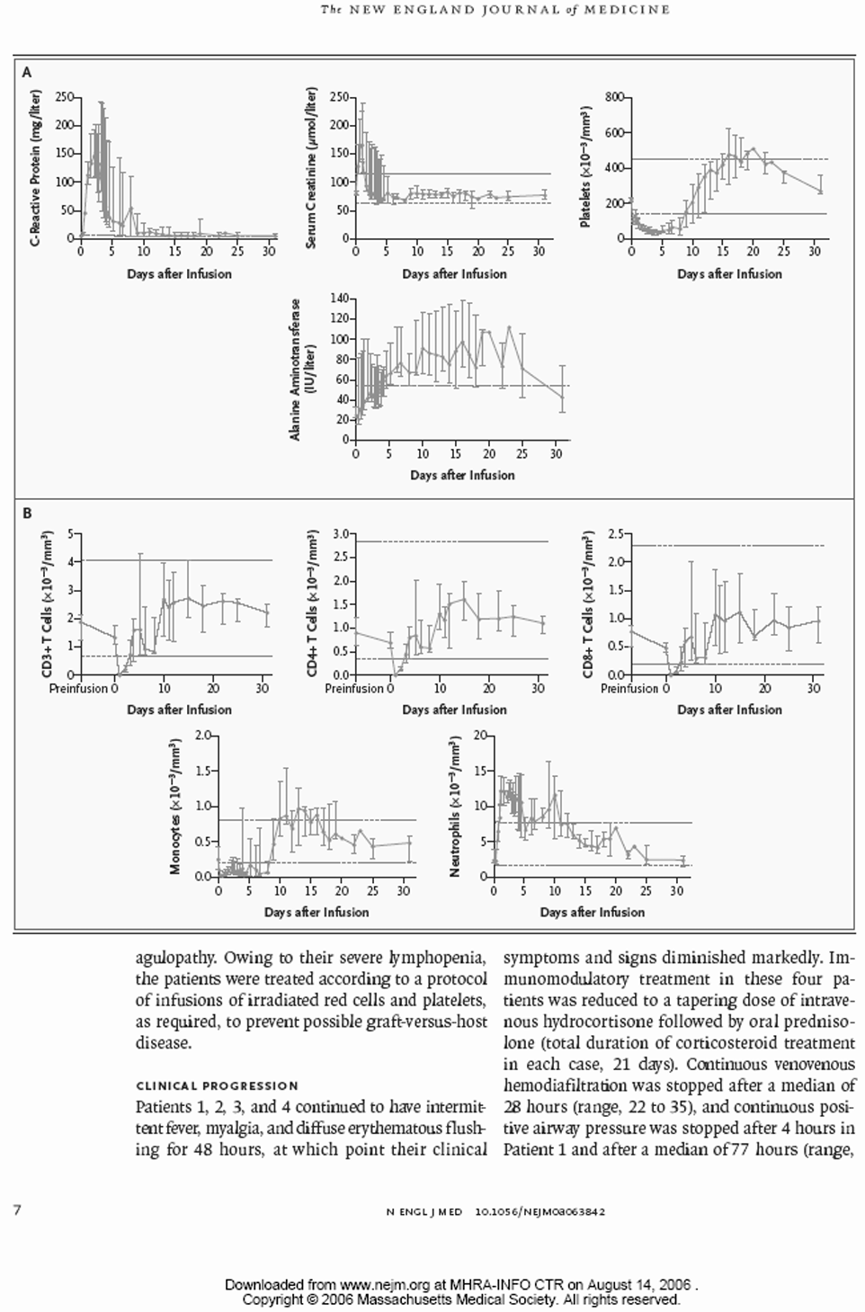
所有细胞因子在第1天和第2天都有显著增加。TNF和干扰素γ尤其在第4小时和第2天时出现了非常大的增加(>500-1000倍)。有趣的是,TNF在所有受试者中在TGN 1412给药后1小时内就急剧上升。到第3天,所有细胞因子都有下降的趋势。
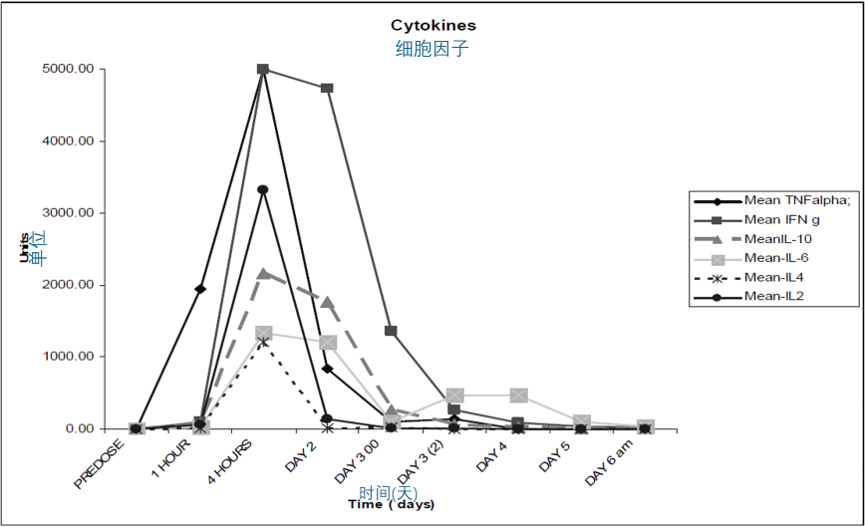
北威克公园医院免疫学实验室的数值这里没有显示。
淋巴细胞亚群;
所有受试者在给予TGN1412后都表现出淋巴细胞减少。整个过程中淋巴细胞水平的比较见下表。
| Blood level (units) 血液水平(单位) | Predose (range)* | T=8 hours (range)* | CTU normal range | T=16 hours (range)** | ICU normal range |
|---|---|---|---|---|---|
| Lymphocytes(x10^9 /L) 淋巴细胞(x10^9 /L) | 1.86 (1.47-2.59) | 0.06 (0.05–0.09) | 1.5–4.0 | 0.04 (0.03-0.07) | 1.10–4.80 |
个体的T淋巴细胞数值变化更大。在所有接受TGN 1412的受试者中,CD3、CD4和CD8细胞都有明显下降。恢复情况也各不相同,CD3细胞的恢复最先,其次是CD4,然后是CD8。CD8细胞的恢复最慢,幅度也最小。
结果;
后遗症
所有受试者在事件发生后都由临床医生随访了三个月。所有人都有残余的限制,尤其是记忆丧失、注意力无法集中和头痛。一名受试者的手指和脚趾出现干性坏疽,仍需要治疗措施。一名受试者有复发性口腔溃疡。
目前还在等待这些个体的血流动力学稳定性、免疫学特征和抗体筛查的进一步数据。
D MHRA GCP、GMP 和 GLP 调查
简介
本部分介绍了药品与医疗产品监管机构(MHRA)针对单克隆抗体TGN1412“首次人体试验”期间发生的严重不良事件所进行的调查。它总结了MHRA和其他欧洲监管机构进行的检查,以及事件发生后对样本进行的测试。本次调查的目的是确定在试验过程中是否存在可能导致严重不良事件的错误。
背景
Parexel(一家合同研究组织)受赞助商TeGenero AG委托,进行单克隆抗体TGN1412的人体初步研究。2006年3月13日,Parexel临床药理研究单位(CPRU)招募了八名健康男性志愿者并对其进行给药。同一天,八名受试者中有六人报告了严重不良事件(SAEs)。
根据Parexel CPRU的说法,受试者出现了“细胞因子释放综合症(Cytokine Release Syndrome)”,被报道为“危及生命(Life-Threatening)”。药物编码由Parexel解封,这确认了六名出现严重不良事件的受试者接受的是"活性药物(active drug)",而两名没有出现不良事件的受试者接受的是安慰剂。
研究设计
一项I期、单中心、双盲、随机、安慰剂对照、单次给药递增剂量研究。四个单次给药剂量:0.1、0.5、2.0 和 5.0 mg/kg,分别在4组各8人中给药。计划总共32名受试者。
首次筛选日期:2006年2月22日
首次给药日期:2006年3月13日
参与试验的主要组织
| 角色 | 名称 | 地理位置 |
|---|---|---|
| 申办方(赞助商) | TeGenero股份公司 | 德国符茨堡 |
| 制造商 | “勃林格殷格翰 [根据 FOIA 第38条已编辑] Parexel” | 德国 |
| GLP 研究 | [根据《信息自由法》第38条已编辑] | |
| 合同研究组织 / I 期临床药理单位 | 英国哈罗,北威克公园医院 |
设施的触发检查
事件发生后,各个设施进行了触发检查。
| 设施 | 检查类型 | 检查日期 |
|---|---|---|
| 德国TeGenero AG | GCP | 2006年3月17日 |
| Parexel,英国哈罗北威克公园医院 | GCP | 2006年3月16日、17日和27日 |
| Parexel | GMP | 2006年3月14日和16日 |
| [根据《信息自由法》第38条已编辑] | GLP | 2006年3月20日-21日 |
| 德国勃林格殷格翰 | GMP | 2006年3月22日-24日 |
检查结果
TeGenero AG 的GCP检查
检查主要关注TeGenero在首次人体研究之前所进行的临床前工作。所有工作都已按照临床试验申请流程的要求提交给监管机构。检查中未发现任何不规范情况。
Parexel 的GCP检查
在检查期间,检查团队对以下区域进行了检查:Parexel临床药理研究单位(CPRU)诊所、招募办公室、接待区、用于存放输液泵的区域、存放盲法信封的办公室、样本实验室和样本存储区。此外,还查看了Clinbase®电子临床试验数据库。
检查员报告称,没有发现可能对接受研究药物的试验受试者所经历的严重不良事件产生影响的问题。
方案遵守情况
在GCP检查中,发现了一些差异:
未遵循文档程序
临床药理研究单位有责任按照方案程序保持适当的记录。Parexel未能以书面形式完成试验受试者的完整病史。一位主要研究者在与一名志愿者口头咨询后,没有将病史文件更新为书面形式。
雇佣程序错误
该"样本库筛查医师(bank screening physician )“入职时未签订有效聘用合同,后续才补签合同。
Parexel的主要研究者未在入职初期,于工作记录中正式授权该样本库筛查医师完整的工作职责范围。在检查过程中,MHRA检查员检查中对该样本库筛查医师进行访谈后,认为其不具备胜任该岗位所需的充分培训经历与从业经验。
保险问题
Parexel有责任审核TeGenero的保险政策,以确保保险已经到位,并且其中没有可能影响本研究志愿者的除外条款。然而,他们未能做到这一点,尽管后来未发现任何此类除外条款。
安慰剂志愿者——未遵守揭盲程序
接受安慰剂的志愿者在完成适当检查以确认其接受的为安慰剂之前,这两个接受安慰剂的志愿者被允许提前退出试验。
合同
在试验开始时,TeGenero 和 Parexel 之间没有合同——随后才签发了一份,而 Parexel 与他们聘请的私人实验室之间只有一份合同草案。
医疗保障
没有正式的系统来提供 24 小时的医疗保障。
Parexel GMP 检查
检查范围包括对设施、设备、质量体系、文档和记录的审查,这些都与单位内 TGN1412 和安慰剂的储存、制备和"放行(release )“相关。
检查过程中没有发现任何不足。
[根据 FOIA 第38条已编辑] GLP 检查
检查的目的是确定为支持 TGN1412-HV 进入人体而进行的关键毒理学研究是否按照 GLP 原则进行。
审计的研究是——[根据 FOIA 第38条已编辑] 研究:“四周的食蟹猴静脉注射毒性研究,观察期为六周”。
此外,还审查了两项验证研究和一项剂量范围研究。
检查员得出结论,该研究是按照 GLP 原则进行的,最终报告中呈现的结果似乎准确反映了原始数据。没有发现重大或关键性缺陷。
似乎非常不可能这些缺陷对参与 Parexel CPRU 北威克公园医院 I 期临床试验的志愿者的状况产生影响。
TGN1412 产品分析测试
事件发生后,独立实验室进行了系列测试,以确定产品是否符合批次放行规格,并进行了额外测试以协助调查事件。测试涵盖了用于毒理学研究的批次(80升批次)以及用于受试者的批次(2000升批次)。
概述
在Parexel CPRU发生严重不良事件后,护理人员已经将试验对象所使用的产品和安慰剂样本收集保管,并被大都会警察调查小组扣押。国家生物标准控制研究所(NIBSC)的工作人员在受控温度条件下将样本运输到位于Potters Bar的NIBSC实验室,并将其转移到温度监控的冰箱中。
产品制造商德国勃林格殷格翰(Boehringer-Ingelheim, BI)提供了参考材料和详细的分析方法,以进行所需的分析。作为英国官方药品控制实验室,NIBSC因其角色被MHRA选中执行产品的大部分分析。然而,NIBSC无法进行的测试则在其他三家实验室进行。[根据FOIA第38条进行了编辑] 这些实验室进行了兔致热原试验、无菌试验和总可生长菌计数。美国俄亥俄州辛辛那提法医化学中心和美国食品药品监督管理局(FDA)进行了毒理筛查测试。MHRA位于泰丁顿的实验室开发并验证了一种适用于检测施用于试验对象的稀释样本中蛋白质和乙酸的方法。
MHRA工作人员根据产品制造商的放行测试和规格制定了检测方案。该测试方案旨在展示最初由BI生产用于毒理学研究的80升批次与用于临床试验的全规模生产批次之间的任何差异。
可供测试的样品
以下测试材料由北威克公园医院的CPRU获得:
冷藏(2 - 8℃)的实验用药物(IMP)TGN1412批号 E5646LO04 样品
冷藏(2 - 8℃)的安慰剂醋酸盐缓冲液批号 5653LO01 样品
以下参考/保留样品由德国 BI 提供:
冻存参考样品 Ref Std DS/1。这些材料是由生产批次 90695 和 90710 合并而成。这是用于毒性研究的 80L 批次所生产的材料。
冻存参考样品 Ref Std DS/2。这些材料来自生产批次 100301-01。这是批号 5646LO04 材料的参考样品。
此外,NIBSC 还储存了更多尚未分析的临床材料样品,以及大都会警察从医生实验室查获的样品。
NIBSC 还储存了实际稀释后的 TGN1412 和安慰剂样品,这些样品是直接用于受试者的。
测试方案
以下测试方案是由MHRA调查团队与NIBSC的分析师共同商定的。该方案基于公司的批次放行规格。然而,在收到公司方法之前,对于SDS-PAGE和尺寸排阻色谱分析,NIBSC使用内部方法进行了分析。这些测试旨在确认分发给试验受试者的研究用药物(IMP)批号E5646LO04符合制造商的放行规格,并与用于原始临床前毒性研究的试点批次(Run 90695/90710)等效。
公司发布的规格测试。
紫外分光光度法检测:- 用于确认IMP批次中总蛋白的浓度,并确保产品中不含不溶性聚集体。
LAL凝胶凝块法检测内毒素:- 用于确认不存在内毒素和致热物质。
SDS-PAGE和等电聚焦:- 用于确认产品与预期的人单克隆抗体(MAb)中发现的蛋白一致,并证明没有产生不良的二聚体或半分子。
尺寸排阻高效液相色谱(SE-HPLC):- 用于确认所获得的色谱图中的主要峰与预期分子量的蛋白一致,并且IMP中的二聚体含量与规格及从试点批次获得的色谱图一致。
BIAcore(细胞结合)检测:- 用于确认产品中含有能够结合所使用CD28构建体的蛋白。
总生菌数和无菌检测:- 用于确认安慰剂和IMP都未被微生物污染。
非公司发布的规格测试。
兔致热原试验:- 用于确认产品不含致热物质。测试的执行还要求产品注射到其他动物种类。(由商业合同研究实验室执行)
异常毒性试验(英国药典):- 测试用于确认IMP TGN1412不含意外的有毒污染物,并且MAb对测试物种无毒性。
毒性筛查:- 使用FDA法医化学中心(FCC)的多种筛查技术进行。测试基于FCC常规执行的以确保美国食品安全和非污染的检测。测试结果将提供最终保证,确认IMP TGN1412未被任何意外的有机或无机毒性物质污染。
获得的结果
所有方法得出的结果都确认,TGN1412 按照给试验对象的方式使用,完全符合"放行(release)“规范。
NIBSC的科学家表示,使用E5646LO04批次和参考材料DS 01(试点规模生产)或参考材料DS 02(批号E5646LO04)得到的结果之间任何细微差异都没有实际意义。
所做分析的结果列在附录K的表1中。
结论
调查显示,不良事件并非由于TGN1412的生产、配方、稀释或给受试者使用过程中出现错误。因此,得出的结论是,该药物在人体中出现了无法预测的生物作用,这很可能是试验受试者出现不良反应的原因。
专家科学小组希望强调,对已给予受试者的药物残余注射器样品的成分和潜在污染尚未进行分析,因此,对于试验受试者的配方、稀释和使用方面的结论,应视为初步结论,需要等分析结果出来后才能最终确定。他们还希望指出,上述临时结论是基于对同一批次TGN1412的分析得出的,该批次已按试验方案处理和稀释,并且与GCP记录推断的操作方式完全一致。对仅给予6名受试者的试验药物可能受到某种形式的污染而给予2名接受安慰剂受试者的药物则未受到污染的这种可能性,这种可能性发生的可能极低。因此,临时结论仍然是合理的。
4.抗CD28单克隆抗体TGN1412
作者、新英格兰医学杂志编辑以及马萨诸塞州医学会已授权转载本文。
本文由在北威克公园医院治疗临床试验志愿者的北威克公园医院医生撰写。





5. NIBSC 特殊细胞因子研究及后续研究
TGN1412 的初步细胞测试
背景
在2006年3月13日,TGN1412这种刺激性CD28单克隆抗体(Mab)在北威克公园医院进行临床试验时,所有六名受试者都发生了严重不良事件。随后进行了初步分析,结果显示该药物符合规格并且似乎达到了临床等级。大部分评估在NIBSC进行,目前已完成。
在与MHRA、ESG主席以及内部讨论后,NIBSC开始了第二阶段调查,旨在探究该分子引发病理反应的机制,并进一步了解其生物学活性。TGN1412注射后不久即出现不良反应,持续数周之久。最初出现的发热、疼痛及因低血压导致的器官衰竭等反应,与细菌内毒素引起的反应一致,尽管在"细菌内毒素(Limulus)“检测中未发现该药物含有内毒素。此外,该药物通过了“经典”兔热原试验,该试验用于检测内毒素和“非内毒素”热原性污染物,但相比体外检测方法,其灵敏度相对较低(1),且已有案例显示兔热原试验未能检出某些后来在人体中引发不良反应的污染物(2,3)。
多年来,针对某些在人体中引发不良反应但细菌内毒素试验或兔热原试验(或两者)均呈阴性的产品,一直采用一种替代的“非药典”检测方法来评估其含有的促炎性和致热性污染物(1, 2, 3,4)。该替代检测方法被不同表述为“体外热原试验”、“单核细胞活化试验”或“细胞因子释放试验”。在此方法中,使用人外周血单核细胞——这些细胞对污染物产生反应时会释放促炎性细胞因子蛋白——与待测药物共同孵育,并定量分析培养基中的一种或多种细胞因子。单核细胞通常以单核细胞成分或稀释后的全血形式进行刺激,检测结果包括肿瘤坏死因子α(TNF-α)、白细胞介素-1β(IL-1β)和白细胞介素-6(IL-6)(5,6)。
由于TGN1412引起的不良反应初期提示释放促炎性细胞因子(如TNF-α、IL-1β和IL-6),因此决定通过两种“细胞因子释放试验”来评估TGN1412的细胞因子释放活性:一种使用从人外周血中提取的外周血单核细胞(PBMC, peripheral blood mononuclear cells),另一种使用稀释的人外周血。所有实验均采用健康志愿者(NIBSC工作人员)新鲜采集的血液。选择PBMC检测系统的原因是,该系统(以IL-6为检测指标)在2002年获得FDA批准后,被制药公司用于终产物释放测试,具有重要的监管意义;此外,该方法已被用于过程中的测试,并成功用于检测一种非内毒素热原(细菌肽聚糖),这种物质曾导致某已获批药物的不良反应(4)。选择稀释全血检测系统的原因在于,该系统包含了在单核细胞纯化过程中丢失的中性粒细胞、血小板和红细胞。与基于PBMC的检测不同,使用全血进行的检测目前仅被验证可用于检测内毒素,尽管此类检测确实具备检测“非内毒素”污染物的能力(8,9)。 在检测细胞因子释放试验中的污染物时,通常要求待测药物本身不具有细胞因子释放活性(7)。但在TGN1412的情况下,不良反应的严重程度和持续时间表明该药物自身可能具有内在的促炎活性,尤其是因为TGN1412在细菌内毒素检测和兔热原检测中均呈阴性。因此,在本研究中,我们采用细胞因子释放试验来检测可能由污染物、或不良生物活性,或两者共同引起的反应。
除了细胞因子释放测试,还开展了实验,以研究TGN1412对淋巴细胞增殖的影响,因为这可能与在北威克公园医院观察到的六名志愿者现象有关。
方法
使用外周血单核细胞(PBMC)进行的测试
NIBSC采用的PBMC细胞因子释放检测方法是一种已与制药行业使用的检测方法实现标准化的方法(7)。PBMC是从不超过四小时的人体肝素化外周血中通过密度梯度离心分离出来的。“测试制备物(the test preparation)“在培养基和供体自身血浆中以每毫升0.5到2.0百万PBMC的浓度进行培养。培养过程使用无菌技术,并且所用试剂和耗材都是无菌且无热原的,在37°C、5% CO₂的湿润空气中培养16到24小时。通过验证的ELISA,将对测试制备物的细胞因子反应与对标准内毒素和适当阳性对照的反应进行比较,这里使用的是CD3刺激单克隆抗体UCHT-1和血凝素PHA。测定指标包括:IL-6、TNF-α、IL-8、组织因子(TF, Tissue Factor)、IL-2、干扰素γ(IFNγ, interferon-γ)(不是所有测试都测定了所有指标)。进行了两次使用PBMC的实验:每次实验测试了来自四个独立供体的PBMC。
使用稀释全血进行测试
将不超过四小时的人的肝素化外周血(最终浓度20%)与供试品一起培养。使用无菌技术以及无菌且无热原的试剂和耗材。血液培养在37±1°C、5% CO₂、潮湿空气中进行16到24小时。使用经过验证的ELISA,将对"测试制备物(the test preparation)“的细胞因子反应与标准内毒素和适当阳性对照(在此为CD3刺激单克隆抗体UCHT-1和植物血凝素PHA)的反应进行比较。读数包括:IL-6、TNF-α、IL-8、组织因子(TF)、IL-2、干扰素γ(IFNγ)。(并非所有测试都测量了所有读数)。使用稀释全血进行了四次实验:每次实验都测试了来自四位不同供体的血液。
在细胞因子释放测试中所测试的TGN1412 的剂量的依据
TGN 1412 以 0.1 mg/kg 的剂量注射给志愿者,而对猴子则使用了远高于此的 50 mg/kg 剂量,即人类剂量的 500 倍。
当剂量为 0.1 mg/kg 时,平均男性体重为 70 kg,血液大约有 5 升。药物溶解在血浆中,血浆约占血液总体积的 60%,所以有效稀释体积为 3 升。
0.1 mg/kg x 70 kg = 7 mg TGN 1412 每 3 升血浆
= 约为2 mg/L血浆
= 约为2 µg/ml 血浆
给予猴子的剂量 = 50 mg/kg = 比给人的剂量多 500 倍。较大的剂量相当于约 1000 µg/ml。
所以,TGN 1412 在细胞因子释放测试中,最终浓度为 1000、500、250、125、62.5、31.25、16、8、4 和 2 µg/ml。
细胞增殖测试
据报道,TGN1412 与 CD28 的相互作用能够诱导增殖刺激信号。为了研究这一点,使用基于流式细胞术的实验方法评估了细胞增殖,该方法采用了“湿法”和“干法”涂覆抗体。评估了在 96 孔板中由 TGN1412 湿法涂覆 10 μg/ml 并在某些孔中风干(每孔 1 和 10 μg)刺激的增殖情况。作为阳性对照,使用了促有丝分裂原 PHA(10 μg/ml)和抗 CD3 单克隆抗体 UCHT-1,涂覆浓度为 10 μg/ml,某些孔风干(每孔 1 μg)。简而言之,将分离得来的人的 PBMC 用荧光膜染料 PKH26 标记,洗涤后以每毫升 1×10⁶ PBMC 三重复板培养。72 小时后,PBMC 用抗人 CD4 FITC 和抗人 CD8 APC 偶联物对PBMC进行反向染色,洗涤后用 2% 体积比的戊二醛固定。将三个重复孔合并,通过 Becton Dickinson FACSCalibur 流式细胞仪采集 30,000 个淋巴细胞。正在增殖的淋巴细胞在分裂时将其细胞膜分配给子细胞,导致荧光染料减半,荧光强度相比母细胞和静息细胞下降。这在流式细胞仪上表现为 PKH26 荧光逐渐减弱的细胞带。使用细胞周期分析软件,将增殖细胞荧光强度的下降用于计算前体频率。用 Cell Quest 软件分析增殖细胞的 CD4/CD8 比例。该方法学以前已有描述 (10,11)。
结果
TGN1412(2 到 1000 µg/ml)与 PBMC(1 百万细胞/ml)孵育 16-24 小时,并没有刺激 IL-6、TNF-α、IL-8 或组织因子(TF)的产生。下面的两个图显示了 IL-6 和 TNF-α 的结果。
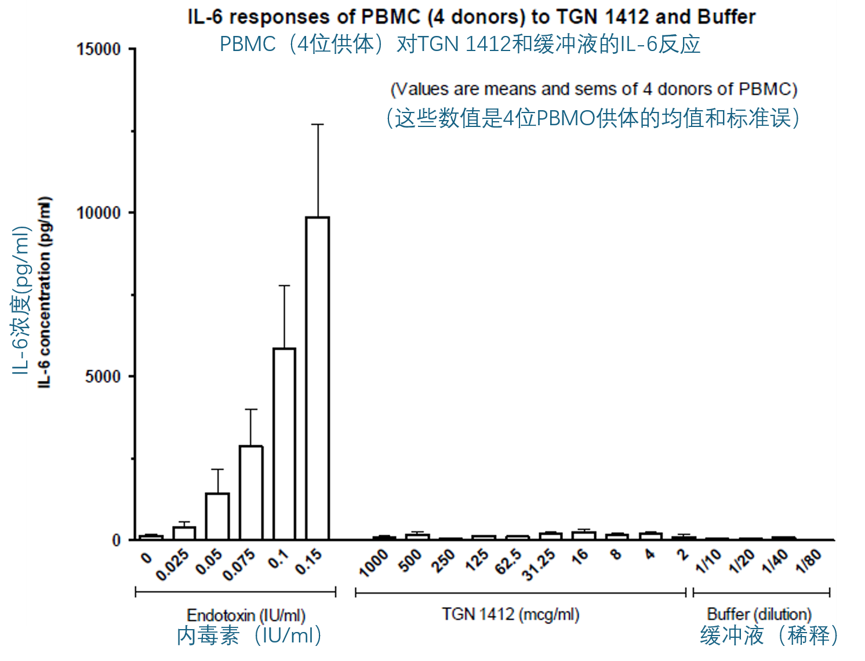
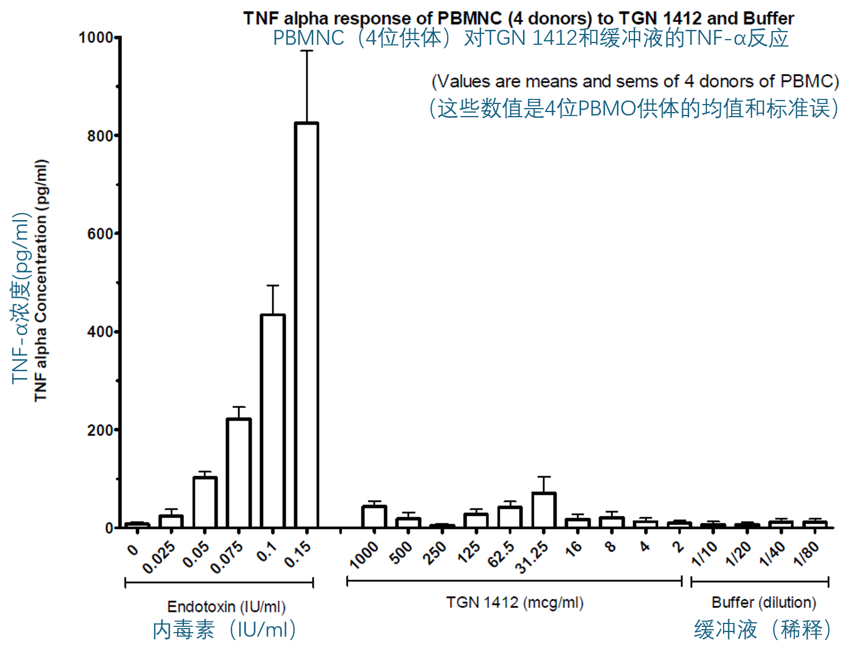
同样地,将 TGN1412(2 到 1000 µg/ml)与稀释后的全血(20% v/v)共孵育 16-24 小时,并没有刺激 IL-6、TNF-α、IL-8 或 IFN-γ 的产生(数据未显示)。在随后对全血进行的实验中,孵育时间延长至 48 或 72 小时,并且选择 IL-2 和 IFN-γ作为读取指标。在这些实验中的一些,所使用的 TGN1412 来自第二个小瓶,并且对该小瓶中的 TGN 再次进行了 Biacore 结合实验,以确认 TGN1412 并未丧失结合 CD28 的能力。第二个小瓶中的 TGN1412 的 Biacore 结果与初次评估结果非常相似,确认了 TGN1412 保持了其生物活性。将 TGN1412(8 到 1000 µg/ml)与稀释后的全血(20% v/v)共孵育 48 或 72 小时,也没有刺激 IL-2 或 IFN-γ的产生。相比之下,CD3 单克隆抗体 UCHT-1(0.5 到 10 µg/ml)和 PHA(2.5 到 20 µg/ml)的阳性对照则刺激了 IFN-γ的释放,而 PHA(2.5 到 20 µg/ml)还能刺激 IL-2 的释放。IFN-γ的反应有些波动,即依赖于供体,但在任何给定的实验中,大多数供体都有反应,虽然对于某些供体,UCHT-1 比 PHA 更有效,而对另一些供体,PHA 又比 UCHT-1 更有效。然而,最显著的观察结果是,仅仅将 TGN1412 与全血或 PBMC 共孵育时,它是无效的。下面的两幅图就说明了这一发现。
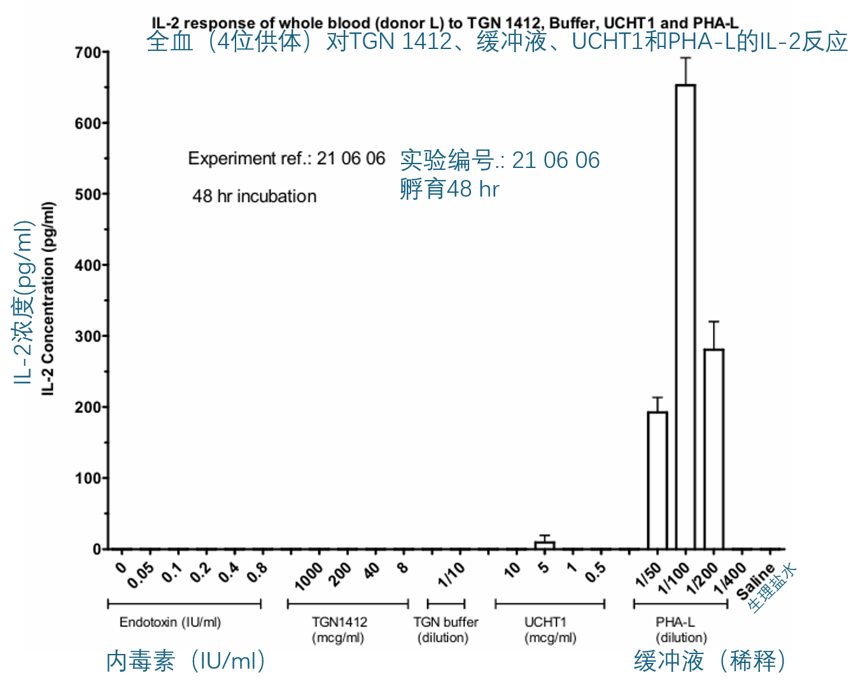
在上述实验中,TGN1412没有效果,这与它在最初用于研究对淋巴细胞增殖的影响的实验中的有效性形成了对比:见下图。在这个实验中,TGN1412被“湿涂”或“干涂”在96孔板的孔壁上。当TGN1412以干涂方式(1 Pg/孔)时会刺激细胞增殖,而湿涂则不会,不过当以10 Pg/孔干涂时,其效果较差。TGN1412(1 Pg/孔)的刺激水平与UCHT-1和PHA的效果相似。虽然这个实验的结果仍在分析中,但进一步的增殖实验已经计划好了,同时还将进行更多细胞因子释放测试,在这些测试中,TGN1412将以“干涂”方式涂在96孔板上,因为这似乎是向细胞呈递药物更有效的方法。

初步实验结论和进一步实验建议
仅仅将TGN1412加入PBMC或稀释血液中并不会刺激“细胞因子风暴”的发生,也不会刺激淋巴细胞增殖。这表明北威克公园志愿者经历的严重不良反应,不能仅仅通过CD28的简单结合导致的细胞因子快速释放来解释。在96孔板孔壁上“干涂”TGN1412可能是一种更有效的方法,这可能是由于交联作用。
应研究将TGN1412添加到包被有抗人IgG4 Fc抗体涂层的板孔中的效果。另外,针对sCD28在人类和食蟹猴血液中可能的作用、TGN1412对其他刺激(如内毒素)反应的影响,以及TGN1412对内皮细胞及内皮- PBMC复合体的影响的实验,可能会提供有关TGN1412的有价值信息。同时,通过流式细胞术检测细胞内细胞因子,并以人体注射剂量进行食蟹猴体内重复给药毒理实验、在食蟹猴中测试的500倍剂量以及中间剂量或不同剂量,也会非常有参考价值。
对TGN1412的细胞实验后续研究
背景
对从北威克公园医院医院回收的未开封瓶子和注射器中的TGN1412进行了分析,该分析于2006年10月完成(请参阅Robin Thorpe提交给MHRA的报告),结果显示该药物符合其规格,且似乎是临床级别的。在与MHRA、ESG主席以及内部讨论之后,第二阶段的调查在NIBSC开始,试图了解导致病理反应的机制,并获取有关该分子生物活性的更多信息。TGN1412的细胞实验中期报告(10 07 06)已提交给ESG。自那时起,NIBSC又进行了进一步的TGN1412细胞实验,其研究结果总结如下。虽然已经学到了很多东西,特别是在如何在体外将TGN1412最有效地呈现给细胞方面,但需要注意的是,这项工作仍在进行中,有些结果需要进一步确认。
方法
体外实验。使用外周血单核细胞(PBMC)的细胞因子释放研究,以及使用流式细胞术进行淋巴细胞激活和增殖的测试,都按照2006年7月10日的中期报告中描述的方法进行,不过TGN1412在呈递给人类或食蟹猴PBMC时用了几种不同的方案:
(1) 作为水溶液
(2) 湿法直接涂在96孔板孔壁上
(3) 干法直接涂在96孔板孔壁上
(4) 作为水溶液,在由人内皮细胞(HUVEC(JR))形成的单层细胞存在下
(5) 作为水溶液,与水溶性抗人Fc抗体一起使用
(6) 湿法涂在已经干法涂在96孔板孔壁上的抗人Fc抗体上
在16-24小时的细胞因子释放研究中,只进行了方案(1)到(4),因为所有测试的抗人Fc抗体都检测到细菌内毒素呈阳性,而细菌内毒素是促炎细胞因子释放的强效刺激物,而且针对从与人内皮细胞共培养中获取的淋巴细胞进行的淋巴细胞激活和增殖测试仍未进行。
和以前一样,包括了合适的阳性和阴性对照:CD3刺激的单克隆抗体UCHT-1、植物血凝素(PHA, phytohaemagglutinin )以及同型匹配(IgG4)但特异性无关的人单克隆抗体,即非抗-CD28抗体。
另外,现在已经进行了进一步的研究,包括在TGN1412刺激3天后测量T淋巴细胞活化标志物的表达,以及在TGN1412刺激6小时后进行的细胞内细胞因子染色,用于检测白细胞介素-2(IL-2)和干扰素γ(IFNγ)。
体内实验。这些实验在12只食蟹猴身上进行,其中两只静脉注射了TGN1412缓冲液(对照组),另外两只分别静脉注射了以下剂量的TGN1412:0.1、0.5、5.0和50 mg/kg,所用TGN1412取自刚开封的药瓶。此外,还有两只动物注射了0.1 mg/kg的TGN1412,这部分药物取自从北威克公园回收的注射器#5。TGN1412是以缓慢静脉注射的方式给药,模拟了在北威克公园试验中尽可能接近的给药速率。选择这些剂量的理由如下:0.1 mg/kg是北威克公园六名志愿者接收的剂量;5.0和50 mg/kg是TeGenero报告的食蟹猴临床前安全性测试中使用的剂量;0.5 mg/kg是为了填补0.1和5.0 mg/kg剂量之间的剂量–反应曲线;注射器#5中含有与最严重不良反应相关的TGN1412样本。
食蟹猴在麻醉状态下接受TGN1412注射。注射后维持麻醉2小时,以便每小时监测血压、心率和核心体温。在此期间采集血液样本进行血液学、血液生化和肝功能测试、细胞因子释放测定及药代动力学研究,从而可以详细监测任何即时不良反应。注射TGN1412 24小时后,对食蟹猴进行麻醉以进行外周淋巴结活检,观察淋巴组织淋巴细胞变化,并采集血液样本进行血液学、血液生化和肝功能测试、细胞因子释放测定及药代动力学研究。第3或第4天再次采集血液样本用于血液学、细胞因子释放测定及药代动力学研究。第7天终止食蟹猴实验,采集血液用于检测,并采集组织进行组织病理学分析。
结果
细胞因子释放研究。
将TGN1412(2.0–1000 µg/ml = 0.25–125 µg/孔)加入水相至PBMC(每孔200,000个细胞)中,并未刺激肿瘤坏死因子α(TNF)、白细胞介素-6(IL-6)和IL-8的释放。同样,将TGN1412湿涂在孔壁上(每孔1 µg)也没有刺激PBMC(每孔125,000个细胞)释放促炎性细胞因子。相反,将TGN1412(每孔1.0和10 µg,但不是0.1 µg)干涂在培养PBMC(每孔125,000个细胞)16-24小时的孔壁上,刺激了TNF、IL-6和IL-8的生成。细胞因子反应的大小比例为IL-8>TNF>IL-6。将TGN1412(0.1、1.0和10 µg/ml)加入水相至PBMC(每孔50,000或125,000个细胞)与人内皮细胞(单层,每孔15,000或68,000个细胞)的共培养体系,培养16-24小时后,刺激了TNF、IL-6和IL-8的生成。细胞因子反应的大小比例为IL-8>TNF=IL-6。
淋巴细胞"激活/活化(activation)“和"增殖(proliferation)“的测试
增殖研究
将用荧光膜染料PKH26标记的人PBMC通过流式细胞仪检测其对不同剂量(0.5µg/ml到1000µg/ml)TGN1412的增殖反应。在水相中、湿涂板上或与抗人Fc抗体交联的TGN1412均未产生显著的增殖反应。只有通过干涂法将TGN1412固定在板上,或者通过抗人Fc抗体捕获后干燥在板上,再洗涤,才能在PBMC与固定的同型对照抗体或TGN1412孵育3天的实验中引起显著的淋巴细胞增殖。对增殖淋巴细胞的表型分析显示,CD4+淋巴细胞是主要的响应群体。增殖数据显示,所有CD4+细胞都进入分裂阶段,最终没有静息的CD4+细胞残留。固定的TGN1412诱导的增殖水平非常高,相当于该实验中作为阳性对照的促分裂原所引起的反应。通过捕获的抗人Fc抗体的TGN1412甚至引发了更强的增殖反应,这表明干涂TGN1412测得的反应可能低估了实际反应的强度。TGN1412刺激淋巴细胞,同时伴随增殖细胞活化标记物CD25的上调。
增殖反应以每10^6淋巴细胞的前体细胞频率进行定量。采用该方法测定TGN1412的增殖反应,发现最佳剂量范围为2到10 µg/ml。在更高剂量(100或1000µg/ml,相当于在食蟹猴中测试的剂量)下,观察到反应低或无增殖。相比之下,2 µg/ml(相当于北威克公园人体给药剂量)可引发强烈的"多克隆(polyclonal)“增殖反应。
经过3天TGN1412刺激后,洗涤并转移到不含TGN1412的新板上的淋巴细胞仍然可以继续增殖3天。
细胞内细胞因子染色
在分泌抑制剂存在下,用固定化的TGN1412刺激人PBMC 6小时后,对IL-2和IFN-γ进行了测定。与对照组相比,CD4阳性淋巴细胞在用固定化TGN1412刺激6小时后产生了显著的IL-2。
IL-2阳性淋巴细胞的水平与本试验中用作强阳性对照的"促有丝分裂原(mitogen)“所观察到的反应相当。在CD4阳性淋巴细胞上检测到固定化TGN1412刺激后细胞内IFN-γ的染色。然而,这一反应远小于促有丝分裂原刺激时的反应,促有丝分裂原可激活CD8阳性淋巴细胞产生比CD4阳性淋巴细胞更多的IFN-γ。固定化TGN1412的剂量–反应关系与增殖反应的剂量–反应关系一致。使用抗Fc抗体捕获的TGN1412时,IL-2阳性淋巴细胞水平甚至更高,这表明用干燥的TGN1412测得的反应可能低估了TGN1412实际可引发的反应强度。
IL-2和IFN-γ的细胞内细胞因子染色反应与用TGN1412刺激的3天培养上清液的ELISA数据一致。也就是说,在TGN1412的小剂量刺激下,IL-2水平高,而IFN-γ水平低;但大剂量时则不明显。
来自食蟹猴的数据
在给予食蟹猴各种剂量的 TGN1412 后,在总体健康状况、血压、心率、体温、血液学、生物化学或肝功能方面均未观察到明显的不良反应。进一步的分析、检测和组织病理学研究仍在进行中。以人体剂量给药的注射器材料也没有引起不良反应。
在初步的体外实验中,用固定化的 TGN1412 刺激食蟹猴的外周血单核细胞(PBMC)时,细胞并未出现增殖反应。早期迹象表明,食蟹猴的 PBMC 会被 TGN1412 “激活(activated )",但不会经历"增殖(proliferation)"。然而,当向培养了固定化 TGN1412 的食蟹猴 PBMC 中加入外源性的人 IL-2 时,观察到了强烈的增殖反应。单独向食蟹猴 PBMC 培养物中加入人 IL-2 并未观察到增殖反应。
人和食蟹猴淋巴细胞对固定化 TGN1412 的体外激活和增殖反应总结
| TGN1412 诱发活化 | TGN1412 诱发增殖 | IL-2 诱发活化 | IL-2 诱发增殖 | TGN1412+IL- 2 诱发活化 | TGN1412+IL-2 诱发增殖 | |
|---|---|---|---|---|---|---|
| 人PBMC | ++++ | ++++ | – | – | 无法测试* | 无法测试* |
| 食蟹猴PBMC | ++ | – | – | – | +++ | +++ |
*: TGN1412 单独使用时会刺激活化、IL-2 分泌和增殖。
结论
本研究显示,只有在以这些研究中确定的有效方式呈现给细胞时,TGN1412才有能力引发细胞因子释放和淋巴细胞增殖。当通过干燥固定在板上、结合内皮细胞或被固定的抗Fc抗体捕获固定时,TGN1412刺激细胞因子释放,包括TNF、IL-2、IL-6、IL-8和IFN-γ,并在体外测定中显著增殖人类CD4淋巴细胞。TGN1412在水相或交联相时未诱发细胞因子释放或增殖反应。这些体外检测数据表明,北威克公园医院试验中给予志愿者TGN1412剂量接近最大免疫刺激剂量。与人类不同,任何剂量下给予食蟹猴TGN1412均未出现严重不良反应,且除非在培养中加入IL-2,否则食蟹猴淋巴细胞在被固定TGN1412刺激时不会增殖。
进一步研究
目前正在进行的实验正在进行,以确定脉冲TGN1412(即先在内皮细胞单层中加入TGN1412,随后用洗去未结合的TGN1412,然后再加入PBMC)是否更有效地刺激细胞因子释放。这可能是由于未结合TGN1412预期会与结合TGN1412竞争,因此在CD28受体处作为结合的TGN1412的竞争性拮抗剂。此外,正在进行人PBMC和人MRC5成纤维细胞的共培养实验,以确定是否可以用除内皮细胞外的单层细胞结合TGN1412使其在体外具有生物活性。这些实验随后将重复进行,在可行范围内,将用食蟹猴PBMC替代人PBMC,以考虑可用于此类研究的少量食蟹猴血液。此外,还将比较TGN1412刺激炎症性细胞因子释放的人类稀释全血与食蟹猴稀释全血。
References
- Duff, G.W. and Atkins E. (1982) The detection of endotoxin by in vitro production of endogenous pyrogen: comparison with Limulus Lysate Gelation.
J. Immunol Methods 52: 323-331, 1982
- Dinarello, C.A., O’Connor, J.V., LoPreste, G., Swift, R.L. (1984). Human leukocyte pyrogen test for detection of pyrogenic material in growth hormone produced by recombinant Escherichia coli. J. Clin. Microbiol, 20, 323-9. 1984
- Poole, S., Thorpe, R., Meager, A., Hubbard, A.R., Gearing, A.J.H. (1988) Detection of pyrogen by cytokine release. Lancet, 8577, 130.
- Martis L et al. Aseptic peritonitis due to peptidoglycan contamination of pharmacopoeia standard dialysis solution. Lancet, 2005, 365(9459): 588-94.
- Poole, S., Gaines Das, R.E. (2001) Towards a ‘human pyrogen test’. J Parenteral Sci. 6(2), 63-4.
- Poole, S., Mistry, Y, Ball, C., Gaines Das, R.E., Opie, L.P., Tucker, G., Patel, M. (2003) A rapid ‘one-plate’ in vitro test for pyrogens. J Immunol Methods, 274, 209-20.
- Gaines Das RE, Brugger P, Patel M, Mistry Y, Poole S. Monocyte activation test for pro-inflammatory and pyrogenic contaminants of parenteral drugs: test design and data analysis. J Immunol Methods, 2004, 288(1-2): 165-77.
- Nakagawa, Y., Maeda, H., Murai, T. (2002) Evaluation of the in vitro pyrogen test system based on proinflammatory cytokine release from human monocytes: comparison with a human whole blood culture test system and with the rabbit pyrogen test. Clin Diagn Lab Immunol, 9, 588-97.
- Hoffmann et al. International validation of novel pyrogen tests based on human monocytoid cells. J Immunol Methods, 2005, 298:161-73.
- Givan AL, Fisher JL, Waugh M, Ernstoff MS, Wallace PK. A flow cytometric method to estimate the precursor frequencies of cells proliferating in response to specific antigens. J Immunol Methods. 1999 Nov 19;230(1-2):99-112.
- Wade-Evans AM, Stott J, Hanke T, Stebbings R, Berry N, Lines J, Sangster R, Silvera P, Walker B, MacManus S, Davis G, Cowie J, Arnold C, Hull R, Almond
N. Specific proliferative T cell responses and antibodies elicited by vaccination with simian immunodeficiency virus Nef do not confer protection against virus challenge. AIDS Res Hum Retroviruses. 2001 Nov 1;17(16):1517-26.
参考文献
- Duff, G.W. 和 Atkins E. (1982) 通过体外内源性致热原产生检测内毒素:与鲎试剂凝胶法的比较。J. Immunol Methods 52: 323-331, 1982
- Dinarello, C.A., O’Connor, J.V., LoPreste, G., Swift, R.L. (1984). 用人白细胞致热原试验检测重组大肠杆菌产生的生长激素中的致热物质。J. Clin. Microbiol, 20, 323-9. 1984
- Poole, S., Thorpe, R., Meager, A., Hubbard, A.R., Gearing, A.J.H. (1988) 通过细胞因子释放检测致热原。Lancet, 8577, 130.
- Martis L 等. 因药典标准透析液的肽聚糖污染导致无菌性腹膜炎。Lancet, 2005, 365(9459): 588-94.
- Poole, S., Gaines Das, R.E. (2001) 迈向“人致热原试验”。J Parenteral Sci. 6(2), 63-4.
- Poole, S., Mistry, Y, Ball, C., Gaines Das, R.E., Opie, L.P., Tucker, G., Patel, M. (2003) 一种快速的“一板式”体外致热原试验。J Immunol Methods, 274, 209-20.
- Gaines Das RE, Brugger P, Patel M, Mistry Y, Poole S. 单核细胞活化试验用于检测注射用药物中的促炎和致热性污染物:试验设计和数据分析。J Immunol Methods, 2004, 288(1-2): 165-77.
- Nakagawa, Y., Maeda, H., Murai, T. (2002) 评估基于人单核细胞促炎细胞因子释放的体外致热原试验系统:与人全血培养试验系统及兔致热原试验比较。Clin Diagn Lab Immunol, 9, 588-97.
- Hoffmann 等. 基于人单核细胞的创新致热原试验的国际验证。J Immunol Methods, 2005, 298:161-73.
- Givan AL, Fisher JL, Waugh M, Ernstoff MS, Wallace PK. 一种流式细胞术方法,用于估算对特定抗原增殖的细胞前体频率。J Immunol Methods. 1999 Nov 19;230(1-2):99-112.
- Wade-Evans AM, Stott J, Hanke T, Stebbings R, Berry N, Lines J, Sangster R, Silvera P, Walker B, MacManus S, Davis G, Cowie J, Arnold C, Hull R, Almond N. 用猿免疫缺陷病毒 Nef 疫苗接种引发的特异性 T 细胞增殖反应和抗体,并不能提供病毒挑战保护。AIDS Res Hum Retroviruses. 2001 Nov 1;17(16):1517-26.
6. 利益相关者咨询总结
向专家科学组的口头陈述
“主席(Chairman )“邀请了广泛的"利益相关者(stakeholders )“提交关于事件的口头或书面意见。
以下利益相关者向主席及专家科学组的代表做了口头陈述。
利益相关者
布赖恩·吉纳里博士,萨里大学临床研究中心主任;
帕特里克·瓦兰斯教授,医学科学院;
英国制药工业协会(ABPI)和生物工业协会(BIA),由科林·多勒里爵士、戴维·契斯韦尔博士、理查德·佩克博士、彼得·劳埃德博士、理查德·巴克博士和艾思琳·伯纳德女士代表;
特里·汉布林教授、马丁·戈尔教授和莫妮卡·普鲁斯博士,代表基因治疗咨询委员会(GTAC);
莎莉·伯特尔斯博士,代表英国癌症研究机构(Cancer Research UK);
马尔科姆·博伊斯博士,人类药理学协会(AHPPI)主席;
J.S. 德·博诺博士,癌症研究院;彼得·韦斯伯格教授,英国心脏基金会;
马克·费尔德曼教授,伦敦帝国学院肯尼迪风湿病学研究所负责人;
莉兹·艾伦博士,合同临床研究协会主席;
罗伯特·霍金斯教授,英国癌症研究机构医学肿瘤学教授,
曼彻斯特克里斯蒂医院。
专家科学小组代表
戈登·达夫教授(主席)、罗伯特·莱克勒教授、莱谢克·博里西维奇爵士、斯蒂芬·英格利斯博士、薇薇安·帕里女士、马克·沃尔波特教授、亚历克斯·马克汉姆教授、妮瓦·海茨教授、安德鲁·麦克迈克尔教授、鲍勃·赫普尔爵士、穆尼尔·皮罗莫哈梅德教授、约翰内斯·勒沃教授、拉尔斯·克拉雷斯科格教授、凯文·帕克教授、赫尔曼·瓦尔德曼教授。
主席对利益相关者的开场致辞
主席欢迎了各位利益相关者,并感谢他们与专家科学小组的成员会面。他解释说,ESG(专家科学小组)独立于MHRA和CHM,将致力于识别具有新药理机制的药物实体的普遍适用的、高度风险的因素。然后,它将尝试将这些风险因素进行规范化,以提出如何优化首次人体临床试验安全性的建议,同时不对有用新药的开发设置不必要的障碍。这一过程的一部分将是采纳来自学术界、工业界和公共领域的广泛利益相关者的意见;这也是为何需要进行“利益相关者咨询(stakeholder consultation)”会议的原因。
主席还解释说,在ESG报告发布之前,所有文件和会议内容都是保密的,发布后,除根据《信息自由法》第38条保留的材料外,所有信息将进入公共领域。
布赖恩·詹纳里博士,萨里大学临床研究中心主任。
詹纳里博士在I期临床试验方面有着丰富的经验,并提到萨里大学临床研究中心为企业进行了大量研究。他表示,TGN 1412事件促使他向专家科学小组表达了自己关于申办方提供给临床单位的数据质量和数量的看法。
他的报告内容是关于早期临床试验阶段的信息需求,虽然他向ESG报告的内容与TGN 1412事件没有直接关联,但对于早期临床开发中受试者的更广泛安全性仍然具有相关性。
报告涵盖了一般原则、试验产品的化学与药学、临床前生物学与安全性测试、临床数据的充分性以及研究者手册的重要性。
帕特里克·瓦兰斯教授,医学科学院。
瓦兰斯教授主持了在TGN 1412事件后成立的医学科学院工作组。科学院此前向一期临床试验专家科学小组提供了一份关于抗体疗法测试的立场文件,这也是他演讲的主题。他认识到科学院无法获取公开领域外的信息,但工作组得出的结论总体上作为此类疗法的最佳实践原则是有效的。
演讲内容涵盖了临床前数据的预测价值、起始剂量的预测、临床地点的考虑以及风险因素的识别。
临床前测试的预测价值包括动物和人体体外及体内数据中效果比例的重要性,以及在判断动物模型的预测价值时,人体和动物细胞体外药物效果的平行比较的重要性。
英国制药工业协会(ABPI)和生物工业协会(BIA),由科林·多勒里爵士、戴维·奇斯韦尔博士、理查德·佩克博士、彼得·劳埃德博士、理查德·巴克博士和艾斯琳·伯纳德女士代表。
奇斯韦尔博士介绍了该小组,并解释说ABPI/BIA团队代表一个大约由20名专家组成的专项工作组,由科林·多勒里爵士和他本人共同主持,研究TGN1412事件的经验教训。他表示,由于并非所有信息都公开,因此没有对TGN1412进行司法调查,而是会为未来的研究提供一份“最佳实践”指南。
他强调,目前关于早期临床开发的审查程序和法规已被证明非常有效,并使此类研究非常安全。TGN1412事件应被视为完全特殊的情况,任何新的建议都不应导致采用清单式的方法。
ABPI/BIA的报告涵盖了生物药物实体的作用机制和代谢、所使用的动物模型及其可预测性、将前临床数据转化为临床应用、试验本身的进行、“生物药(biologicals)“的生产与控制、方案和监管审查,以及科学家和医生在安全性评估方面的教育与培训。
特里·哈姆布林教授、马丁·戈尔教授和莫妮卡·普鲁斯博士,代表基因治疗咨询委员会(GTAC)。
戈尔教授在方案审查方面有丰富的经验,并且多年来一直关注单克隆抗体的开发。哈姆布林教授在免疫疗法领域有长期经验,自1974年起就开始用抗体治疗患者。他在会上展示了他在1994年对单一受试患者进行的一项使用三特异性抗CD3/CD2/CD28抗体研究的未发表数据。
这次报告主要涵盖两个方面,首先是人体剂量,包括健康志愿者与患者,以及1994年进行的首个三特异性抗CD3/CD2/CD28抗体的人体研究。该抗体的效果与TGN1412的效果有相似之处。
莎莉·伯特尔斯博士,代表英国癌症研究机构(Cancer Research UK)
伯特尔斯博士负责英国癌症研究机构的早期临床试验和临床前开发,并向ESG通报了该机构在推进新的癌症治疗进入人体实验方面非常活跃。伯特尔斯博士介绍了CRUK的项目组合,指出其中有9个为生物制品,其中一些属于专家科学小组的职权范围。
她的报告内容涉及从临床前研究到首个人体试验的转化,以及这些试验的设计。涵盖的内容包括总体经验、专业知识和经验、减少动物使用、独立专家审查研究和数据、肿瘤药物开发、风险收益分析、人体转化以及安全性考虑。
马尔科姆·博伊斯博士,制药行业人体药理学协会(AHPPI)主席,
博伊斯博士是一家一期临床试验单位的临床主任,专注于临床药理学。他同时也是制药行业人体药理学协会(AHPPI)的主席。AHPPI成立于1988年,拥有162名会员,其中大多数大型制药公司和合同研究机构(包括派瑞赛尔)都有代表。AHPPI的主要目的是向会员提供持续的临床药理学教育。
博伊斯博士做了题为《新的药物物质在人体的首次给药》的演讲。演讲内容涵盖了英国的一期临床单位、试验发起方、试验类型、体内大分子与小分子、试验前的考虑事项、工作人员培训、相关方之间的交流、一期临床试验的既往安全性、健康参与者与患者、剂量和研究设计的选择,以及审查与评估。
J. S. de Bono 博士,癌症研究所
他的演讲题为“癌症的一期临床试验”,涵盖了被认为是关键的问题,包括癌症一期试验的回顾、抗体治疗、抗体一期试验、风险最小化——益处最大化,以及癌症药物的志愿者研究。
他强调,对于癌症研究,使用健康志愿者或癌症患者的原因是不同的,而且近年来新型治疗的获益机会有所增加。抗体治疗被认为是癌症治疗中的重大成功,这也带来了显著的治疗进展。
彼得·韦斯伯格教授,英国心脏基金会
韦斯伯格教授告诉ESG,英国心脏基金会(BHF)支持各种研究,包括临床试验;然而,他们并不委托研究。他表示,BHF的立场是以患者安全为首要任务,并支持在I期临床试验中尽量降低受试者的潜在风险。
他认为,尽快从临床试验中发生的不良事件中吸取经验非常重要,并建议将不良事件的试验数据提交到一个中央点(例如监管机构),然后将信息传达给相关各方。
马克·费尔德曼教授 FRS(伦敦帝国学院肯尼迪风湿病研究所所长)
费尔德曼教授做了题为“特异物种分子试验设计”的报告。他介绍了肯尼迪研究所在类风湿关节炎中开发和使用抗TNF-α单克隆抗体的经验,以及他们降低风险的方法。费尔德曼教授在报告最后讨论了改善新型特异物种疗法测试的方法,包括广泛的体外分析和使用转基因小鼠。
莉兹·艾伦博士(合同临床研究协会主席)
艾伦博士与B·霍尔特博士一同出席,并进行了题为《“生物药(biologics )“和新靶点新化合物一期研究的考虑事项》的演讲。
与会成员了解到,合同临床研究协会(CCRA)是一个拥有28个成员的行业组织,其中9个参与一期研究。
CCRA认为,在对具有特定靶点的"生物药(biologics )“和新化合物进行临床前评审时,安全性是最重要的,需要特别关注作用机制、潜在的靶点效应后果,以及动物模型的可预测性和同源性。
会议中指出,英国一期临床试验的安全性非常好,而TGN 1412事件是前所未有的。还提供了数据显示,从1992年到2000年,81,471名受试者中有171例发生了严重不良事件(0.21%)。
罗伯特·霍金斯教授(英国癌症研究基金会医学肿瘤学教授,曼彻斯特克里斯蒂医院)
霍金斯教授做了一个题为“癌症免疫疗法的临床开发”的演讲,他分享了自己在癌症疫苗、靶向超抗原和基因改造T细胞方面的工作经验。
口头报告及相关讨论的完整细节已在附录C的会议完整记录中呈现。
利益相关方向专家科学小组提交的书面意见
以下利益相关方提供了书面意见:
ABPI / BIA,早期临床试验工作组联合报告,
皇家内科医学院药学学会,
纳菲尔德生物伦理委员会(Sandy Thomas 教授,主任),
Ravinder Maini 爵士教授,肯尼迪风湿病研究所,ICL,
Camilo Colaco 博士,ImmunoBiology Ltd,剑桥
国家癌症研究所,消费者联络组(Roger Wilson 先生(主席)),
皇家学会(David Read 教授,生物学秘书及副主席),
Lynda Wright 博士,专业监管事务组织执行董事
Kate Webb,高级政策顾问,Which。
皇家统计学会,Tim Holt 教授,
Focus on Alternatives,Gill Langley 博士,
J Robinson Associates,
Datamonitor, Press release。
ABPI / BIA,早期临床试验工作组联合报告
提交了一份完整的报告,涵盖了事件背景、作用机制和生物活性、安全性评估的物种/动物模型选择、临床前数据向临床的转化、临床试验的执行、生产、方案和监管审查,以及教育和培训。这些领域的调查结果和建议都进行了讨论,并提出了首次在人体使用新型生物制药时需要考虑的要点。
皇家内科医师学院药物医学学院
这篇文章集中讨论了临床试验前信息的审查、试验中采用的临床操作、申办方和审查者的责任,以及参与临床试验的工作人员的培训和能力。
纽菲尔德生物伦理委员会(Sandy Thomas 教授,主任)
这篇文章调查了知情同意的问题和给予试验参与者的诱因,同时也涉及到动物研究的有效性以及寻找替代动物使用的方法。
Ravinder Maini 爵士教授,剑桥大学肯尼迪风湿病研究所
文中提供的评论包括动物模型预测毒性作用的能力、识别毒性作用的能力以及迅速且适当反应的必要性。文中还指出,首次人体试验应由了解目标生物学及预期反应的临床医生来进行。
卡米洛·科拉科博士,ImmunoBiology有限公司,剑桥
这篇文章发表于《科学家》5月5日的期刊中,作者的建议是开发一种机制,让外部专家评审小组可以与MHRA联系,提供及时、相关且保密的专业意见。
国家癌症研究所,消费者联络小组(主席:罗杰·威尔逊先生)
这份提交中涉及的问题包括知情同意流程、动物研究、标准操作程序、参与者选择、同时用药以及对以往研究产生的数据的分析。
皇家学会(大卫·里德教授 FRS,生物学秘书兼副会长)
本文关注于三个具体领域:具有新作用机制的生物分子和针对特定物种的全新药物、新型针对免疫靶点的药物,以及使用动物进行临床前测试的有效性。
琳达·赖特博士,执行董事,专业监管事务组织(TOPRA)
TOPRA的回应包括对该组织的背景介绍,以及概述其认为在临床试验和更广泛教育领域可以做出的贡献。
凯特·韦伯,高级政策顾问,Which
Which的回应文件以一般评论、临床试验方案、试验管理、试验监管以及结论和建议为标题进行呈现。
英国皇家统计学会,Tim Holt 教授,
学会成立了一个工作小组,审查“首次人体”研究的统计设计考虑,特别是针对单克隆抗体以及更广泛的新型生物制品和生物技术类别。
关注替代方案,Gill Langley 博士,
书面提交材料包括关于动物实验系统性弱点的看法、一种更安全临床研究的方法,以及对新范式的需求和相关结论。
J Robinson Associates,
没有给予在报告中发布的许可。
Datamonitor, Press release。
对生物制品推动制药行业增长的独立评论。
书面提交材料及相关文件的完整细节见附录 C 和 D。
7. 预测从临床前到临床转化中的风险
背景
TGN1412 试验已经得到两家欧洲监管机构和当地研究伦理委员会的批准,但所有接受抗体的受试者在给药后不久都经历了无法预料且非常严重的副作用。目前还在等待进一步的实验室测试结果,但到目前为止,没有证据表明观察到的严重副作用是由抗体以外的因素引起的,很可能就是抗体的‘靶向作用(on-target)’在试验志愿者体内触发了细胞因子释放综合征。
据我们所知,在所有试验受试者中出现像 TGN1412 那样严重的不良反应是史无前例的。这凸显了迫切需要审查首次人体试验中新药的安全性,以及检视目前药物开发中风险是如何被申办方、研究者和监管机构评估和最小化的。
TGN1412 已经按照现行法规要求进行了临床前测试,但从临床前研究中并未预测出首次人体试验的安全起始剂量。在 TGN1412 的临床前开发阶段进行的几项实验室测试,也由国家生物标准与控制研究所(NIBSC)重复进行了,结果相似。
不过,需要注意的是,在NIBSC进行的进一步测试显示,有可能在体外创造出让TGN1412引起人血细胞释放细胞因子和T细胞增殖的条件。将抗体干燥涂在塑料表面固定,或者使用固定的第二抗体,或者在血管内皮细胞单层存在的情况下让人血细胞暴露于TGN1412,都能导致细胞因子释放和/或T细胞增殖。
值得注意的是,到目前为止,还没有在体外重复出对食蟹猴T细胞增殖的类似刺激作用,而且食蟹猴在体内广泛剂量范围内使用TGN1412时也没有出现不良反应。
这些研究补充了关于不良反应潜在机制的信息,也解释了为什么在人体和动物细胞以及动物模型中进行的临床前测试未能预测在试验中给予的 TGN1412 起始剂量对人体的反应。近期在 NIBSC 进行的工作结果,如果得到确认和进一步发展,可能会指出应在未来临床前测试中采用的不同方法,特别是针对具有细胞表面靶点的激动性(刺激性)抗体。
ESG 的目标是明确从 TGN1412 经历中可以学到什么,并考虑这些发现能在多大程度上推广,以提高未来高风险新药首次人体试验中志愿者的安全性。在我们的职权范围内,我们研究了是否以及如何从药物的性质、药理靶点及其预期接受者中识别潜在风险。
我们得出结论,某些新药本身的特性以及它们作用的分子靶点中存在可识别的危害。识别这些危害对于首次人体临床试验的安全性尤其重要。下面的结论总结了ESG的讨论,并考虑了从利益相关者收到的书面和口头意见。
在首次人体试验中,新药的毒性可能很早就出现,即在给药后不久,或者较晚出现,即在一段时间后才显现。ESG关注的是发生得早或立即发生且可能危及生命的严重急性毒性。对于晚期毒性的监测需求及监测时间长度,则最好根据个案来决定,需要考虑药物类型、其药理靶点及体内半衰期等因素。
我们并没有尝试以任何正式的方式量化或排名风险,我们使用“高风险药物”这个术语是指那些在我们职权范围内可以识别出风险因素的药物。
我们的许多建议涉及影响首次人体试验安全性和价值的决策,我们认为这些决策必须逐案做出,并且要有科学依据。我们意思是,开发者应为每个个案提供决策的理由,而监管者应评估这些理由,同时考虑试验志愿者的安全以及将获得的信息。
这些建议旨在适用于参考范围内对高风险药物的人类首次接触,但有些建议可能适用于一期临床试验整体。
某些新药的固有危险
作用的物种特异性
有些药物,尤其是生物药物,比如为了与人体细胞受体有亲和力而设计的重组细胞因子或多肽激素,以及针对人体目标抗原筛选的单克隆抗体,可能会表现出很强的物种特异性。
一般来说,针对人体特定分子目标的药物,在其他物种中(如小鼠、大鼠和兔子)活性可能大幅降低,但在分子结构更接近人体的非人灵长类中仍可能有一定活性。
除了分子对目标分子的亲和力差异外,其他因素也会影响实验动物的生物反应,无论是数量上还是质量上,与人类的反应相比。例如,分子靶点在组织中的分布可能不同,靶点结合的细胞后果可能不同,细胞调控机制可能不同,代谢途径可能不同,或者对初始生理扰动的补偿反应可能不同。
对于新的药物,如果体外研究显示其在人体细胞与实验动物细胞之间存在物种特异性作用,那么实验动物体内反应在预测人体体内反应方面的价值将显著降低。
这个问题可能更容易出现在生物药物中,尽管任何类型的药物,包括小分子化学药物,如果对人类靶点高度特异,也可能表现出物种特异性的作用。
在实际操作中,这意味着使用相对物种特异性的药物进行的临床前动物研究:
x 可能无法再现人类的预期药理效果;
x 可能给出误导性的药代动力学和药效动力学结果;
x 可能无法反映“靶向作用(on-target)”毒性。
此外,大型与人类兼容的生物分子很可能在其他物种中引起免疫反应,而且由于其免疫原性,动物研究可能无法预测重复给药此类药物实体的效果。
一种药物实体的物种特异性并不意味着首次人体试验中总是有更高的风险,但它确实使在动物实验中进行风险的临床前评估变得更加困难,有时甚至可能不可能。因此,需要采取高度谨慎的做法。
还需要注意的是,在体外人类和动物细胞中出现类似反应,并不一定保证体内反应也会相似。
在许多新药临床前开发的方面,考虑到“三个R”(改善、减少和替代动物使用)的前提下,动物研究仍然是必要的,包括测试“非靶向作用(off-target)”和“靶向作用(on-target)”毒性,以及理解与新药及其靶分子相关的人体基础生物学。大多数新药,甚至可以说几乎所有新药,都是通过精心设计的动物研究获得的生物学见解产生的。我们想强调的关键点是:在新药开发的临床前阶段,必须决定从动物研究中可以学到什么,以及预测人体反应和剂量-反应关系时可能存在的限制。
新型药物实体及作用机制
对于首次人体临床试验来说,具有非常新颖作用机制的药物,其不可预测的不良反应风险通常更大,因为在临床前开发和向临床开发过渡的过程中没有以往经验可供参考。
如果药物还存在物种特异性,那么从体外人类细胞或组织的研究来预测人体体内反应就显得尤为重要。
新型药物实体及其新作用机制可能涉及药物的物理特性、其靶向对象以及预期的生物反应。这些因素都可能受到体内环境的影响,例如药物的酶促或非酶促修饰、药物在体内的分布及其在作用部位的持续时间、靶点的数量和细胞分布、无法预料的下游事件(包括细胞或生化通路放大作用,这些在体外模型中未出现过),以及代偿性稳态机制的影响。
激动机制
许多新药物实体被设计成通过以前未被用作治疗靶点的细胞受体起作用。细胞表面的受体可以专门识别通常称为配体的细胞外分子。配体与受体的相互作用会引发细胞反应,这种反应通常通过生化信号通路传递到细胞核。在很多情况下,细胞核中的反应表现为基因活性调控的变化。一些受体,例如类固醇激素受体,位于细胞内部,因此没有从细胞表面开始的信号通路。
针对细胞受体的新药物可能会模拟天然配体的作用,或者占据受体位置而不引发细胞反应,但阻止天然配体与受体互动。在受体层面上,前者称为受体"激动剂(agonist)",后者称为受体"拮抗剂(antagonist)"。
药物可以对抑制细胞活动的受体以及刺激细胞活动的受体产生激动或拮抗作用。因此,受体激动剂和拮抗剂可以根据它们所作用的受体的生理功能,刺激或抑制细胞。
TGN1412 被设计成针对 T 淋巴细胞上的 CD28 受体的超激动剂,这是一种在正常生理中起辅助刺激作用的受体,会导致 T 细胞活化,同时增加细胞因子产生和细胞增殖速度。
通常来说,那些能激动细胞、引起细胞活化和增殖的药物可能风险更高,尤其是它们的作用靶点位于生物放大级联反应中的时候(见下文)。
“效力(Potency)”
新型药物实体可能被设计成比它们要模拟的天然配体具有更强的作用。
例如,这可能通过以下方式实现:
x 与受体的亲和力更高,或与天然配体相比占据更多受体;
x 对目标受体的信号作用比天然配体更强;
x 多价性,可以交联多个目标受体。
一些药物的作用也可能会由于药代动力学特性而被放大,这些特性会导致目标细胞暴露于药物的程度高于天然配体。
TGN1412 的设计是与 CD28 分子上不同于该受体天然配体的部位相互作用,从而直接刺激细胞信号传导,而无需像自然中那样通过 T 细胞抗原受体提供辅刺激。因此,它具有一种新的作用方式。
TGN1412 绕过了正常的控制机制,这种机制需要抗原刺激才能激活免疫反应,它只扩增携带适当抗原受体的 T 细胞克隆,而 TGN1412 则独立于抗原特异性刺激了许多 T 细胞克隆。
TGN1412 是一种高分子量单克隆抗体,不能通过肾脏排出体外,因此在体内的半衰期以周计算。作为一种抗体,TGN1412 一旦注射后很难从体内清除。它会进入含有许多用于抵御感染的抗体的免疫球蛋白池。
“多功能(Multifunctional )“分子
抗体是多功能分子的一个好例子。它们是复杂的蛋白质,能够高度特异性地识别三维分子结构。虽然它们的自然功能是识别微生物病原体并帮助消除它们,但抗体的分子识别特性也可以在开发新药中加以利用,从而识别特定的分子靶点。
如果靶点是可溶性介质,抗体可能会结合它并中和其作用,就像用于治疗类风湿性关节炎的抗TNF抗体一样。如果靶点是细胞表面受体,抗体可以作为拮抗剂阻断它,或者作为激动剂刺激它。某些类型的抗体与细胞结合还可能通过激活细胞毒性蛋白(补体)和其他机制导致细胞死亡。
由于抗体有两个识别抗原的臂,从理论上讲,一个抗体可能同时结合两个细胞表面受体,从而大大增加细胞的信号反应。还有一种可能性是,针对细胞受体的抗体可能会结合那些已经从细胞膜上切割或释放的受体,形成可溶性免疫复合物。没有实验研究的话,这种情况的功能后果无法预测。不过,如果人体和测试动物之间在受体脱落方面存在差异,那么即使抗体对靶点的结合亲和力相似,动物试验的结果也可能无法很好地反映人体的反应。
抗体的一个主要部分,区别于抗原识别区域,也可以通过与各种不同类型的细胞上的细胞受体结合而产生重要的生物学效应。抗体的这一部分被称为Fc部分,其细胞表面受体被称为Fc受体。通过其抗原识别部分与细胞表面受体结合,以及通过Fc部分与不同细胞上的Fc受体结合,抗体可以充当桥梁,将不同的细胞靠近,有可能进一步激活这两类交叉连接的细胞。尽管IgG4(TGN1412的特殊抗体类型)的Fc受体被认为很少,但根据目前的证据,不能排除TGN1412在体内具有这种交叉连接活性。
某些类型药理靶点的固有风险
总体来说,对于任何可能对关键生命系统造成严重生理干扰的药物,在首次人体接触时都需要高度谨慎。
免疫系统靶点
免疫系统能够识别微生物和异常细胞,并生成淋巴细胞克隆来清除它们。免疫系统的初始激活会触发一个自我放大的淋巴细胞增殖和可溶性介质释放的级联反应,以产生免疫反应。
通常,这种反应仅限于由其抗原受体特异性决定的一小部分淋巴细胞克隆,通过这些受体传递初始激活信号。
作用于免疫反应早期阶段的药理药物实体,其效果在体内可能因免疫级联的激活而被大大放大。
当药物实体是靶向激活淋巴细胞受体的激动剂,且通过旁路使多克隆淋巴细胞被激活时,免疫系统过度刺激的潜在风险会增加。类似的考虑也可能适用于那些在关闭免疫反应的调控通路中起作用的拮抗剂。
其他具有潜在生物放大作用的靶点
上面已经确定了一些针对免疫系统的危险因素,但药理学刺激其他自我放大的级联系统(如炎症反应和血栓-止血通路)可能也存在固有风险。
风险因素总结
ESG得出的结论是,由于与创新药物相关的固有科学不确定性,不可能列出与研究性药物及其靶点相关的所有可能风险因素的完整清单。因此,对风险或危害的评估必须基于科学,并按个案进行。
不过,有一些因素在从临床前到临床的过渡中显然可能带来更高的风险。这些因素包括:
x 任何可能对关键身体系统造成严重生理干扰的药物;
x 激动或刺激作用;
x 新型药物和作用机制,缺乏先前经验;
x 药物的物种特异性,使临床前风险评估困难或不可能;
x 药物的效力,例如与天然配体相比;
x 多功能药物,例如双价抗体、Fc受体结合结构域;
x 与细胞相关的靶点;
x 绕过正常控制机制的靶点;
x 免疫系统靶点;
x 在体内可能产生大幅生物放大的系统中的靶点。
除了在首次人体临床试验中应该考虑的可识别因素外,还有其他一些因素需要考虑,其中有些未来可能会变得更加重要。例如:
x 靶点在动物模型中不存在的药物;
x 联合产品,例如基因治疗中的逆转录病毒载体(即使组件之前在人体中使用过,新组合也可能带来新的风险);
x 与原研产品有显著差异的‘生物类似物’,例如在翻译后修饰上有所不同;
x 具有新型佐剂的疫苗,尤其是那些将抗原引导到免疫细胞表面信号分子上的,或者旨在刺激淋巴细胞或抗原呈递细胞产生促炎细胞因子的疫苗;
x 药物实体的给药途径、剂量方案或"制剂(formulation )“的重大改变。
我们的职权范围列出了三类在首次人体接触中可能被认为先天具有更高危害风险的药物实体,或者在临床前风险评估可能更困难的药物实体。这三类分别是:
x 具有新作用机制的生物分子。
x 高度物种特异性的新品试剂。
x 针对免疫系统的新试剂。
虽然我们对谨慎用药的建议主要针对这三类药物中的任何一种,但我们也意识到,如果通过仔细的科学风险评估可以判断首次人体接触的潜在危害较低,可能会有所例外。
尽管上面提到的一些风险因素示例在"生物药药物实体(biological agents)“中更常见,但它们也可能适用于小分子药物。对可能增加首次人体试验风险的因素以及需要更加谨慎的情况的描述,并不是一个完整或全面的清单,仅作为示例提供。
显然,适用于某个药物及其药理靶点的潜在风险因素越多,从临床前到临床开发的风险就越高,需要越谨慎,特别是在物种特异性是一个因素时。
8. 风险降低与风险管理
ESG的总体目标
我们的目标是回顾并总结从TGN 1412试验中可以学到的经验,这次试验在人类中的安全起始剂量无法从已完成的临床前研究中预测。我们考虑了如何在不阻碍创新或增加开发有用新药不必要障碍的前提下,优化未来“首次人体”试验的安全性。
我们的建议是在考虑ESG职责范围以及可能增加风险的普遍特征的基础上制定的。虽然我们没有尝试以任何正式方式量化或排名风险,但我们使用“高风险药物”一词是指那些符合我们职责范围,并且存在可能增加首次人体试验志愿者危害风险的因素的药物。
我们的许多建议涉及影响首次人体试验安全性和价值的决策,我们认为这些决策应根据具体案例进行科学合理的处理。我们的意思是,开发者应为此类决策提供明确的理由,而监管机构应评估这些理由,同时考虑试验志愿者的安全以及试验将获得的信息。
关于提高首次人体高风险药物试验安全性的观点,是ESG在讨论的总结性结论,这些讨论考虑了自2006年7月26日发布中期报告前后从利益相关者那里收到的所有书面和口头意见。
背景
目前的临床前开发和首次人体研究指南是从几十年的低分子化学药物经验中发展而来的,并已被调整以适用于以前的生物药物类型,主要是经过改良或天然的生物产品,如疫苗和从人类及动物来源提取的产品。这些指南以前的药物类型和一期临床试验中表现良好,TGN1412试验之前的安全记录也不错。然而,许多新型药物,如TGN1412,是先进生物技术的创新产品,可能需要不同的方法。
第一代生物技术药物,包括治疗性单克隆抗体、重组细胞因子、干扰素和激素,在癌症、自身免疫、炎症性疾病以及许多其他消耗性非恶性疾病的治疗领域取得了真正的临床进展,并为患者带来了巨大益处。我们可以预期,也希望在未来看到这些新药物的大量增加,以及其他新型生物药物,例如基于核酸的药物,用于疾病的治疗和预防。
相比于更简单的化学实体,这些药物实体可以设计出更高的特异性和效力,这也为临床前和临床开发带来了挑战。现在显而易见,这类药物的临床前开发不能依赖于曾经对小分子化药或前几代生物药物有效的方法。
有许多潜在的新药类别,每个类别固有的风险可能会大不相同,尽管一些概括性的总结是可能的。由于风险存在差异,不太可能有任何单一的、主要的风险降低技术或方法可以充分优化所有类型高风险新药的首次人体试验的安全性。
ESG(专家小组)得出的结论是,从多种来源整合信息,通过迭代过程,是优化某些类型新型生物药首次人体试验中志愿者安全性的最佳方法。类似的考虑也适用于特定物种的小分子药物,当动物研究中‘靶向’毒性的检测可能不可靠时,也需要类似的处理。这同样可能适用于意外的“非靶向”毒性,这可能揭示新的生物学机制。
在药物开发的各个方面采用多层次的风险降低和风险管理方法应当成为常态,尽管在开发的不同阶段(如临床前和临床阶段)有操作上的区分,但应保持对整个流程的整体概览,并随着新信息的积累不断完善。
临床前研究
决定是否开发一种新药,应基于对未满足临床需求的识别以及有力的科学理由证明其治疗效益。一般来说,与药物类型及其药理靶点相关的潜在风险越高,其开发背后的科学和临床依据就越重要。
通过动物模型的研究,可以了解新药的预期药理反应及其可能的不良反应。然而,需要理解动物模型对人体反应的预测价值,尤其是当药物或其体内靶分子已知或怀疑存在特异性效应时。
对于高度新颖的药物实体,监管科学家与相关研究领域的独立科学家之间进行讨论是非常重要的,以评估这一新型药物实体的完整生物学效应。关于从"整体动物(whole animal)“研究和使用人类及动物组织的体外实验中可以有效获得哪些信息的决定,必须基于科学,根据不同技术获得信息的价值及其与体内人类相关性的情况来做出。
例如:
o 使用体外的细胞和组织来评估在人体和动物模型物种中靶分子结合亲和力的可比性。
o 评估可测量的细胞或组织对受体占据的反应在形态或生化变化方面的可比性,包括预先规定的反应以及通过筛选程序(如RNA或蛋白质谱)可能检测到的细胞变化。目的是测试人体和模型物种生物反应的相似性。应进行定性和定量比较。
o 在某些新药高度物种特异的情况下,特别是像抗体或细胞因子这样的生物药物,使用与人类药物同源的动物模型来测试生物反应可能会很有价值。这可以增加对靶点生理作用的理解,但不能必然预测人体的反应。
o 使用转基因动物细胞、组织或整体动物模型,或细胞嵌合体来评估对人类特异性药物的反应,也可能具有价值。
o 新药在动物和人类细胞或组织中的反应之间的相关性,以及体外与体内动物反应之间的关系,可能通过迭代过程逐步优化预测人体体内反应的能力。
o 试验设计应由适当的统计方法指导,以最大化试验信息价值并尽量减少受试者风险。可能可以开发新的统计方法或计算机建模来优化临床前信息对人体首次使用试验的预测价值。参与首例人体临床试验设计的统计学家应充分了解临床前开发阶段所有可能影响受试者安全的相关因素,以及期望通过试验获得的新信息目标。
最终,新药的临床前开发策略,以及用于收集优化首例人体试验安全性信息的实验方法,都是基于科学的决策,必须由有能力且经过适当培训的研究人员根据具体情况逐案做出。
严格遵循基于现有药物经验制定的指南,可能会在处理新药类别时带来虚假的安全感,也可能在特定情况下过度依赖不适用的方法。
关于新药物临床前开发策略的决策,以及用于收集与首次人体临床试验安全性相关信息的实验方法,必须基于科学,由具有适当培训的个人逐案做出并提供理由。
新药物的临床前开发由国际公认的指南规范,特别是 ICH S6 和 M3(M3 (R1) 《药物进行人体临床试验的非临床安全性研究》CPMP/ICH/286/95,S6 《生物技术衍生产品的临床前安全性评估》CPMP/ICH/302/95)。
监管机构应考虑这些指南对高风险药物的适用性,尤其是首次用于人体时。应考虑制定更明确的指导。最好的方式可能是在欧盟层面先出台初步指导,然后以此为基础在 ICH 层面提出建议。
最近在NIBSC进行的实验结果表明,在体外研究中可以采取不同且有信息量的方法,这些方法可能更能预测体内反应。对于高风险的药物实体,临床前开发计划应确保对该药物实体的生物活性和效力有充分了解。
x 对于基于创新技术的高风险药物实体和先进药品的首次人体试验,监管流程应定期进行审查。
随着当前生物学、生物化学、信息学和生物技术的进展,我们可以预期将鉴定出许多新的治疗靶点,并开发满足临床未满足需求的创新药物。重要的是,药品的监管应以科学为基础,监管流程需要跟上科学和技术的发展,并适应未来药物的产生。
信息共享
临床前研究
尝试收集与首次人体试验中受试者安全相关的未发表临床前信息似乎非常必要,这有助于为未来使用类似药物的试验提供规划参考。英国或欧洲的数据库可以与其他国家的数据库相连,形成一个全球性的数据库。有迹象表明,制药和生物技术行业以及研究资助机构可能愿意以这种方式探索信息共享,而来自其他领域的研究人员,如学术中心和研究机构,也可能会做出贡献。
药物开发者、研究资助机构和监管机构应该加快收集与人类暴露安全相关的未发表临床前研究信息。第一步应重点关注那些显示使用高风险药物或药物组可能对人体有潜在危险的临床前报告。这可以为欧盟和国际层面的监管机构之间的信息共享提供平台,例如以机密数据库的形式。强烈鼓励研究者将这些数据提交到数据库中。
我们认为最终目标应该是一个开放获取的数据库,这一可行性需要进一步探索。为了避免延迟分享关键安全信息的风险,那些原本不会公开的研究报告可以先存放在仅全球监管机构可访问的安全数据库中,并定期审查延迟开放获取的原因。
一期临床试验
在2004年《欧洲临床试验指令》实施之前,在英国,研究人员若要在健康志愿者身上开展一期试验,而该试验对志愿者没有潜在治疗益处,是不需要向监管机构申请许可的。一些其他欧洲国家及其他地区可能也存在类似情况。
尚不清楚在2004年前英国进行了多少首次人体试验,也不清楚其结果如何。在之前研究一项药物过程中获得的有用信息,并不总是公开的,也不一定能被正在进行相同或相似药物试验的其他人获取。
ESG 在一次利益相关者咨询中听到了一个具体例子:一项针对三种靶点(包括 TGN1412 的靶点)的抗淋巴细胞抗体的一期临床试验,其与人体安全性相关的信息从未被公布。
临床试验结果未发表的原因可能有很多,包括负面结果较难被科学期刊接受发表、商业机密问题,或者项目在早期开发阶段就被放弃。
新药的开发者和监管机构都应该考虑如何鼓励并加快将首次人体临床试验的过去信息,以及未来可获得的信息,收集到数据库中的方式。应该探索对这些信息开放访问的可行性。例如,访问该数据库会对研究伦理委员会有帮助。
监管机构应考虑如何加快在欧盟及全球范围内在监管者之间共享首次人体临床试验的安全信息。这当然应包括关于高风险药物首次人体试验的经验信息。安全性结果为阴性的试验也应包括在内。该数据库的范围可能会扩大,包括目前可能不被认为是高风险的产品,或者在开发后期进行的试验,这些试验可能对类似产品首次人体使用提出强烈警告。
在欧盟,这种信息的收集和共享可以基于现有的临床试验数据库模型,比如自2004年以来的首次人体试验的EudraCT数据库,以及用于‘疑似意外严重不良反应(SUSARs)’的EudraVigilance数据库。2004年前的首次人体试验的相关信息可以自愿提交。这样可以确保国家监管机构获取到相关的安全信息。
自2004年以来,英国要求在一期临床试验中,对怀疑的意外严重不良反应(SUSARs)必须向监管机构报告。欧盟所有国家也有相同的规定,其他国家也有类似的安排。在欧洲的意图是各监管机构之间共享这些信息,但我们建议,这应该包括全球监管机构之间的信息共享。同时,还应探讨开放获取这些数据的可行性。
从临床前到临床开发的转化
在首次人体试验中计算起始剂量是影响受试者安全的一个关键因素。TGN1412的临床前研究未能预测出适合人体试验的安全起始剂量,目前所有可用证据都表明给出的剂量太高。过去对小分子药物有效的剂量计算指南可能并不适用于大分子生物药物,尤其是在药物作用具有物种特异性时,从动物研究预测人体安全剂量可能很困难甚至不可能。
通常,对于小分子药物,可以以在动物中没有效果(未观察到效果水平,NOEL)或没有不良反应(未观察到不良反应水平,NOAEL)的低剂量作为起点,然后计算出具有高安全余量的剂量作为首次人体试验的起始剂量。一般来说,药物的物种特异性越强,动物研究信息作为人体起始剂量参考的可靠性就越低。
为了解决这个问题,应该采取一种更广泛的剂量计算方法,基于所有相关信息。例如,这可能包括考虑药物的新颖性及其作用机制、药物的物种特异性程度、人类和动物细胞的生物学效应剂量反应曲线,以及相关体内动物研究的剂量反应数据。
对于受体配体,还需要测量或计算受体占有率与浓度的关系,以及体内靶点和靶细胞的暴露程度。一般来说,即使受体占有率与生物学反应之间的关系未知,首次人体暴露的剂量也应计算出在目标处产生低受体占有率的浓度。
有利益相关者在一份提交材料中建议,生物药物首次人体试验的起始剂量应该设定为能达到人体剂量反应曲线最低端的剂量。这个剂量可以通过人体受体占用情况和细胞剂量反应研究来估算,再结合合格动物模型的信息以及类似药物的经验。这将给出一个“最小预期生物学作用水平”(MABEL),起始剂量应设在这个水平以下。ESG认为,这是计算首次人体试验安全剂量的一个非常有用的方法。
将物理化学和生物学研究的信息结合对类似药物的已有知识,很可能提高高风险药物首次人体试验的安全性,并且如果能够更好地获取以前针对类似药物的临床前和一期研究结果,这种方法还可以得到进一步提升。
ESG总体的意见是,当临床前信息可能由于任何原因无法很好地预测人体体内的反应时,起始剂量应计算得偏向谨慎一方。
有人认为,如果给出的剂量过低而无法提供任何有用信息,可能会质疑试验的伦理性。然而,ESG的意见是,由于事先很难预测一个低到无效的剂量,在合适的剂量递增方案下使用非常低的起始剂量,可以在不造成严重实际障碍的情况下提高首次人体试验的安全性。
有人认为,对于高风险药物,可以在指定的低剂量水平下给药,例如微克或纳克级别(‘微剂量’、‘纳剂量’),作为一般性指导可能是有价值的。但对于某些效力极高的药物,这些指定剂量仍可能对首次人体试验来说过高。ESG认为,虽然这些指导有助于在剂量计算上引起注意,但‘经验法则’不如对所有相关信息进行逐案详细评估来得有价值。
还有人指出,在利益相关者的意见中,非常小的亚药理剂量虽然降低了受试者风险,但在剂量递增过程中达到药理剂量时,并不一定能降低风险。对于那些在人类体内剂量-反应曲线非常陡峭的药物,这一点尤其可能成立。
x 对于那些在动物模型中无法证明其主要药理作用能实现预期治疗效果的新药物,应给予特别关注。任何此类试验的剂量选择依据应包括对拟议作用机制的明确理由,同时也要说明该物质在其预期临床用途中的安全性和有效性。
x 剂量计算应采取更广泛的方法,不仅仅依赖于动物研究中的‘未观察到效应水平’或‘未观察到不良效应水平’。初始剂量的计算应利用所有相关信息。需要考虑的因素包括:药物的新颖性、生物效力及作用机制、药物的物种特异性、生物效应在人体和动物细胞中的剂量-反应曲线、体内动物研究的剂量-反应数据、药代动力学和药效动力学建模、靶点占位与浓度的计算,以及人体体内靶点或靶细胞的暴露量计算。
‘最小预期生物效应剂量(MABEL)’方法是实现该目的的一种有效模型。(参见ABPI/BIA报告及相关利益方提交材料)
x 如果不同方法对人体安全剂量的估计值不同,应在第一次人体试验中取最低值作为起始剂量,并在实际起始剂量计算中引入"安全余量(margin of safety)"。
x 当临床前信息因任何原因可能无法很好地指导人体体内反应时,首次人体试验的起始剂量应谨慎计算,以安全为先。后续剂量增加也应谨慎,因为初始剂量可能特别低,并且可能存在陡峭的剂量-反应曲线。
x 应仔细考虑首次人体试验中首剂的给药途径和速度,并密切监测可能出现的过度反应。
例如,在第一次人体接触通过静脉给药的高风险药物时,几个小时的缓慢输注可能比几分钟的缓慢推注更合适。这可以在出现不良反应时进行监测,并在临床需要时停止输注。
试验设计,以及关于受试者人数、起始剂量和剂量递增方案的决策,应基于对药物性质、体内靶点以及预期受试者相关风险的详细评估。正如许多涉及新型高风险药物从临床前到临床过渡的问题一样,这些最终都是基于科学的决策,必须逐个个案慎重做出,并应基于所有相关信息能够证明其合理性。
x 试验设计,包括受试者人数、起始剂量和剂量递增方案的决定,应根据具体情况逐案制定,并且应具有科学和统计上的合理性,同时考虑所有相关信息。
(见来自英国皇家统计学会的利益相关方提交资料)。
试验受试者的"次序(Sequential )“给药
一般来说,首次人体试验中的新药物应该按"次序(Sequential )“给受试者使用,并在受试者之间保持适当的间隔,以限制可能受到严重不良反应影响的人数。给第一位、第二位以及后续受试者用药之间的观察间隔应该根据可能出现的不良反应类型来确定,这取决于药物的性质、作用靶点以及预期的受体。
虽然在人类中不会有可用的药代动力学数据,但通过动物研究仍可能对药代动力学进行一些预测,例如对于那些不会在肾脏中过滤的大型生物分子。
对于首次人体试验的高风险药物,ESG讨论过在给第一个受试者用药后应进行的最低观察期,但也承认,在给第一个和后续受试者用药之间的观察期必须考虑药物本身、其作用靶点、受试者以及可能的不良反应范围等因素。
不同受试者用药之间的监测期应根据具体情况决定。一期临床试验中有关于逐步增加剂量并监测不良反应的方案。对首次使用递增剂量的受试者延长监测期也能提高安全性。
x 首次人体试验的新药物实体应按"次序(Sequential )“给受试者使用,在给每位受试者用药之间应有适当的观察时间。连续给药的观察间隔应根据可能出现的不良反应类型来确定,这些反应取决于药物的性质、作用靶点和受试者,以及药物的潜在药物代谢动力学和"药物效应动力学/毒性动力学(pharmaco/toxico-dynamics)"。不同个体之间以及同一个体的不同剂量之间都应有合适的间隔,并且都应基于现有证据加以说明。
x 在剂量递增过程中,受试者的连续给药之间也应有类似的监测时间。(如上)
首次人体一期试验的受试者选择
一期试验的目的不是评估新药物实体的治疗效果,受试者通常也不会从中获得任何益处。一般来说,是否在健康志愿者或患者志愿者中进行一期段试验,应根据具体情况决定。
需要考虑几个因素,比如所测试的药物实体类型及其分子靶点的固有风险,健康受试者和患者中是否存在该靶点,在健康受试者或患者中可能获得信息的价值,以及这些信息能够推广的程度。
在癌症领域,有在具有高毒性潜力的细胞毒性药物上进行临床试验的历史。通常的做法是先在患者志愿者中进行首次人体试验,这可以确保目标药物作用的靶点存在,同时‘靶向’和‘非靶向’的毒性效应都能被检测到。
尽管在一期试验中预计对受试者没有治疗性益处,但在对高风险、针对严重疾病的药物进行‘风险与收益’评估时,患者志愿者可能比健康志愿者更适合作为受试者,尤其是在所有现有治疗方案已经用尽的情况下。这假设如果患者在首次人体临床试验中获得任何益处,试验药物可以在试验结束后继续用于治疗。
然而,在某些情况下,健康的志愿者可能更适合作为一期临床试验的受试者。例如,当患者同时使用的药物会导致结果解读困难时。
x 是否在健康志愿者还是患者志愿患者中进行首次人体试验的决定,应当经过仔细考虑并充分论证,考虑所有与受试者安全以及可能获得的科学信息价值相关的因素。
最重要的因素始终应该是志愿者的安全、权利和福祉,无论他们是患者还是健康个体,以及从临床试验中可以学到的价值。
首次人体试验的临床方面和环境
“首席研究者(PI, Principal Investigator)“必须具备适当的资格来进行和监督试验。PI 应该确认已经获得了有关被测试药物性质的完整信息、与人体安全相关的临床前开发细节,以及试验设计和方案的合理性。如果有疑问,PI 应该寻求进一步的信息。用于计算起始剂量的方法以及剂量递增策略的科学依据也应提供给 PI。主要研究者应始终确保自己能够做出有根据的临床判断。
在存在可预测的人类严重不良反应风险的情况下,应事先考虑治疗策略。这应包括在有特效解毒剂时的可用性,以及明确的支持性治疗计划,包括重症监护病房(ITU)设施的可用性。
首次人体试验应在适当的临床环境中进行,由具备相应培训和专业知识的人员监督,并且了解试验药物、其作用靶点及作用机制。必须能够立即使用用于医疗急救(如心脏急症、过敏反应、细胞因子释放综合征、抽搐、低血压)的设施,以及用于急性紧急情况稳定患者的设施,同时重症监护病房设施也应随时可用。
x负责首次人体试验受试者照护的首席研究者应始终具备适当的资格,并确保自己对试验药物、其作用靶点和作用机制有足够了解,以便能够做出有根据的临床判断。
应鼓励建立首席研究者国家专业认证体系,以管理进行首次人体临床试验的人员。
x在有可预见的某些严重不良反应风险的首次人体研究中,应事先考虑治疗策略。这应包括在存在特定解毒剂时的可用性,以及清晰的支持性治疗计划,包括事先安排好的重症监护单元(ITU)应急可用性。
x高风险药物的首次人体研究应始终在适当的临床环境中进行,由具备适当培训和专业知识的人员监督,并能立即使用急救和稳定患者的设施,同时事先安排好重症监护单元(ITU)的应急可用性。
在首次人体试验中,人员配备应始终充足,如果志愿者需要过夜,也应有足够的24小时值班保障。所有进行此类试验的临床机构都应有应急情况的标准操作程序,工作人员应通过定期演练保持执行这些程序的专业技能。试验受试者应始终被明确告知,如果在临床试验期间或之后出现不良反应症状,应如何处理。
监管考量
药品的监管,包括临床试验的开展和安全性,应当以药物研发背后的科学为基础。近年来,分子生物学和生物技术的进步带来了新型的高效且具有临床价值的药物。随着生命科学发展速度的加快,在相对短的时间内可能出现重大突破,而且这种趋势可能会持续到未来。因此,监管者必须能够掌握新兴科学领域的广泛专业知识。
监管部门获取独立建议的途径
专业的科学和医学评估人员应该得到与各自领域前沿研究保持联系的外部专家的支持,以确保监管决策基于最先进的科学。
当需要的时候,比如在评估首次人体一期临床试验申请时,监管机构应能够寻求这种支持,例如针对前面讨论过的高风险药物实体的试验申请,这些风险因素包括药物实体的性质、新颖程度、预期的药理靶点以及预期的受试者。
可以对现有的科学顾问结构进行改进,以便向监管者提供高度专业的专长。例如,人类药物委员会的专家顾问小组(EAG)可能承担这一角色,任命由相关专家组成的核心成员,并根据需要增补其他专家。
目前,对于临床试验授权申请的评估规定,因为对流程施加了严格的时间限制,所以在寻求外部意见时可能会遇到一定的后勤困难。设立常设 EAG 的优势在于可以与监管者建立良好的沟通,并能在短时间内提供回应。
如果能够鼓励开发者和监管机构在申请提交之前的更早阶段进行更多沟通,“逻辑方面的问题(The logistical problem)“应该会显著减少,尤其是对于高风险药物。这也可以让监管机构在必要时更早获得外部专家意见。
增加监管机构与研究伦理委员会在高风险药物试验申请方面的沟通机会,也有助于总体安全环境的提升。
虽然通过更慢的监管流程引入不必要的药物开发延迟不符合患者利益,但在极少见的复杂情况下,应考虑对临床试验评估的时间表引入一定的灵活性。
x 强烈建议药品开发者在提交申请之前,尤其是在处理高风险药物时,与监管机构进行更多的沟通,以确保有足够时间适当考虑任何安全问题,同时不会给产品开发带来不必要的延迟。还应考虑增加监管机构与研究伦理委员会之间的沟通方式。
x 对于高风险药物的首次人体试验申请(根据药物性质、新颖程度、预期药理目标和预期受试者来定义),监管机构应该能够获得来自各自领域具有研究经验的独立专家的额外意见。
x 药物委员会的专家咨询小组(EAG)或类似机构可以承担这一角色,核心成员由适当的专家组成,并根据需要吸纳额外专家。
x 对于异常复杂的特殊情况下,应该考虑在临床试验评价时间上引入一定的灵活性。
未来需求
技能和培训
在英国,许多首例人体一期临床试验,可能是大多数,目前都是在与NHS医院和大学无关的商业临床研究机构(CRO)进行的。
这意味着英国的医学院、护理学院、牙科学院和药学院,以及NHS,获得开发和教授下一代一期临床试验专家所需技能的机会正在减少。这对英国一期临床试验的未来以及临床试验总体发展都有负面影响。其他国家的情况可能类似。
如果学术机构和NHS机构与商业CRO建立联系,临床试验规划和实施中的“实践操作”经验的提供可能会增加。大学或NHS的研究生培训项目可以在CRO中安排实习期。另一种在NHS和大学内保留和发展临床试验技能的方法可能是鼓励在它们内部成立一期临床试验专项中心(见下文)。
在英国医学科学院2005年的《更安全的药物》报告(http://www.acmedsci.ac.uk/p102.html)中,强调了培训新一代医生掌握评估药物安全性和有效性所需技能的必要性。在ABPI最近的一项研究报告《维持制药和生物制药行业技能储备》中也发现了类似的需求。(http://www.abpi.org.uk/Details.asp?ProductID=285)
这需要高等教育资助机构和院校、NHS 以及产业之间的合作。鉴于主要利益相关者都同意需要通过新一代临床药理学家及相关专业人员来填补这一技能缺口,这个问题应该是可以解决的,我们也鼓励这样做,以保障未来首次人体临床试验的安全性。
x实际参与临床试验计划和执行的机会应当扩大,例如通过在研究生培训项目中派驻到商业机构的实习期,或者在 NHS 和大学内建立专业中心(见下一条建议)。
随着支持创新药物的科学变得越来越复杂,我们可以预期未来会有越来越先进的药品。例如,这些产品可能非常具体地针对信号通路或核酸序列,并可能包含遗传元素、细胞元素以及先进的物理或生物递送系统。
这些创新产品在欧盟被称为先进疗法药品,被认为在当前临床需求存在的疾病中具有巨大的治疗潜力。但更有效药物的承诺也意味着不可预测的风险。当创新产品进入首个人体试验时,开发能够预见和识别这些风险的技能非常重要。应探索在大学和NHS内建立专门中心的可行性,以开展高风险和先进药品的一期临床试验。
x 应该探索建立专门中心,以进行高风险药物和先进医疗产品的一期临床试验的可行性。
应鼓励开发一套针对进行首次人体高风险药物研究的临床中心的全国性检查和认证系统。虽然我们提到了大学和NHS,但认证应向所有符合规定标准的公私部门中心开放。这将提升培训机会和技能基础。
9. 建议的范围和总结
ESG 的目标是定义从 TGN1412 试验中可以学到什么,并考虑如何在 ESG 的职权范围内,将这些发现推广到未来更高风险新药志愿者试验的安全优化。在本报告中,我们提出了 22 项建议,我们相信这些建议将有助于优化未来首次人体一期段临床试验的安全性,同时不会对有用新药的开发设置不合理的障碍。
我们考虑了可以提高安全性的前临床、临床及监管方面的内容,无论是现在还是未来。建议主要针对高风险药物的首次人体试验,这些药物的风险由其性质、作用方式、药理靶点以及预期受试者决定,但其中一些建议也可能适用于一般的一期段临床试验。
我们的建议旨在补充现行的监管指南,并在涉及新型药物时优化其适用性,尤其是在首次人体试验中可能出现更高毒性风险的情况下。
我们的一些建议涉及的决策,我们认为在临床前开发和临床转化过程中最好采用逐案评估的方式做出。我们建议,这类决策应当提供合理的依据,我们的意思是,药品开发者应提供说明,监管机构应仔细评估,并在必要时参考独立专家的意见。
建议的适用范围
适用于什么类型的临床试验?
这些建议主要适用于“首次人体试验”,而不是一般的一期段试验(后者可能包括对已有人体安全记录的药物进行的试验)。在首次给人类使用高风险药物,且剂量可能产生药理作用时,需要特别小心。
不过,当将可能带来高风险的药物用于一个全新的群体时,也应该格外谨慎,无论这个群体是健康志愿者还是患者。制定首次人体试验的指南时,应考虑这些概念在从一个人群转向另一个人群时的适用性。
ICH E8 指南《临床试验一般考虑(CPMP/ICH/291/95)》(http://www.emea.europa.eu/pdfs/human/ich/029195en.pdf)提供了关于临床试验的一般指导,并在第 3 节中包含了对一期试验的国际公认描述,以及从药物发现到上市的开发阶段的良好概览。
适用于什么类型的药物实体?
我们的权限涵盖三类药物,这些药物在首次人体"暴露(exposures)“时可能对志愿者带来更高的伤害风险,或者在临床前开发中风险可能更难评估。具体类别如下:
x 具有新作用机制的生物分子;
x 高度特异于某些物种的新药物实体;
x 以免疫系统为靶点的新药物实体。
我们希望我们的建议适用于这三类药物中的任何一种,除非经过仔细评估药物的性质、靶分子的生理作用以及预期使用者的情况后,确认首次人体暴露的风险较低。
我们并不建议所有属于上述三类的药物实体在首次人体暴露时一定会带来高风险,但在评估风险被认为较低时,应该进行彻底的风险评估,并提供清晰的科学依据。
例如,常规疫苗虽然旨在刺激免疫反应,但可能并不会带来高风险;或者类似于人体已有安全使用记录的新药物实体,且针对已知靶点,其药理作用可以有把握地预测,也可能不会带来高风险。
我们在本报告中讨论了在首次人体暴露新药物实体时应提高警惕的一些因素。包括:
x任何可能对重要身体系统造成严重生理干扰的因子;
x激动或刺激作用;
x新的药物实体和新颖的作用机制,且没有先前经验;
x特异性导致动物模型中的临床前风险评估困难或不可能;
x药物实体的"效力(potency)",例如与天然配体的比较;
x多功能的药物实体,例如二价抗体、FcR结合域;
x细胞相关靶点;
x靶点绕过常规控制机制;
x免疫系统靶点;
x靶点在具有体内大规模生物扩增潜力的系统中。
在首次人体试验之前,应该始终进行彻底的风险评估。风险评估应在试验文件中清楚说明,并由监管机构全面审查。如果存在重大疑虑,应始终假定风险较高。(见报告第7节)
建议汇总
在临床试验中,受试者的安全、权利和福祉,无论是患者还是健康志愿者,都必须始终是首要考虑因素。我们提出了22条建议,我们认为这些建议将提高未来涉及首次人体暴露于潜在高风险药物的临床试验中志愿者的安全性,这些药物按照我们的职权范围分类,并在报告的第8节中进行了讨论。
这些建议包括:
x 临床前和早期临床阶段的开发;
x 临床试验申请的准备和审查过程,以及为监管机构和申办方提供的早期咨询机会;
x 确定和管理人体起始剂量;
x 首次人体试验的临床环境;
x 培养技能和培训以满足未来需求。
重点是分享与安全相关的信息、首次剂量的计算和管理,以及在评估首次人体暴露于我们职责范围内所述类型的新药试验申请时,获得独立专家意见的监管途径,这类试验具有较高的潜在风险。
每条建议的理由以及我们的意图说明在第8节【译者注:此处作者存在笔误,应为第7节】“从临床前到临床转化的风险预测”和第9节【译者注:此处作者存在笔误,应为第8节】“风险降低与风险管理”中都有概述。建议应结合这些章节来考虑。
利益相关者提出了几个不在我们职责范围内的关注点。这些包括知情同意的流程和信息的清晰度、临床研究者与临床试验受试者在试验前和试验期间的沟通、保险覆盖情况、研究伦理委员会的作用,以及对经历不良反应的试验受试者的临床随访。虽然这些不在我们的职责范围内,但我们认为这些更广泛的问题都非常重要,建议将它们作为高优先事项来处理,并详细考虑。
我们的建议是针对英国当局和英国首例人体试验的申办方提出的,但我们认为重要的是应在欧盟及国际层面寻求共识,以确保全球"临床试验参与者(clinical trial participants)“获得同等保护。
最终建议
临床前和早期临床开发:
- 关于新药临床前开发的策略,以及用于收集与首次人体试验安全性相关信息的实验方法的决定,必须基于科学,由具有"适当培训(appropriate training)“的个人逐案做出并"提供理由(justified)"。
新药的临床前开发是由国际公认的指南来规范的,特别是《ICH S6 生物技术衍生产品的临床前安全性评价》和《ICH M3 (R1) 药物开展人体临床试验的非临床安全性研究》(见下方进一步阅读)。
监管机构应考虑这些指南对于高风险药物,尤其是首次用于人体的药物的适用性。在当前背景下,应提出制定更具体指南的建议。这最好通过先在欧盟层面发布初步指南,然后以此为平台向国际层面提出建议来实现。
- 对于高风险药物实体和基于创新技术的先进药物的人体首次试验,其监管流程应该定期审查。
随着生物学、生物化学、信息学和生物技术的不断进步,我们可以预期会发现许多新的治疗靶点,并开发出满足未被满足临床需求的创新药物。药品监管必须以科学为基础,其监管流程也要跟上科学和技术的发展,并适应这些将带来未来药物的科学与技术。
- 药物开发者、研究资助机构和监管部门应加快收集与人体暴露安全相关的未发表临床前研究信息。第一步应聚焦于那些显示使用高风险药物或药物组合可能对人体造成危险的临床前研究报告。这将为欧盟及国际级别的监管部门之间的信息共享提供平台,例如以机密数据库的形式。强烈鼓励"研究者(investigators )“将这些数据提交到数据库中。
为了安全起见,我们认为最终目标应是一个开放访问的数据库,这一可行性需要进一步探讨。然而,为了避免延迟分享关键信息的风险,本应不公开的研究报告最初可以存储在仅全球监管人员可访问的安全数据库中,并需定期审查延迟开放的理由。
- 监管机构应考虑加快欧盟及全球监管机构之间一期临床试验安全性信息的共享。这当然应包括首次进入人体使用高风险药物的经验。应纳入具有负面安全性结果的试验。这个数据库可能会扩大,以包括目前可能不被视为高风险的产品,或在开发后期进行的试验,这些纳入的试验数据对首次使用类似产品提出了强烈警告。
在欧盟,这种信息收集和共享可以基于现有临床试验数据库EudraCT(自2004年以来的首次人体试验)和EudraVigilance数据库(疑似意外严重不良反应(SUSARs)的模型(见下文)。2004年前首次人体试验的相关信息可自愿提交。这将确保国家监管机构能够获取相关的安全信息。
这需要与制药行业对话,确定哪些研究报告应发布以及何时发布,但由于行业已承诺公开已上市药物的临床试验结果,并鼓励申办方发布可能存在重要安全隐患的开发失败结果,这应是可行的。监管机构应探讨开放访问这些数据的可行性。
SUSARs是疑似意外严重不良反应。它们之所以意外,是因为它们的性质或严重程度与适用的产品信息不符(例如,未经批准的研究产品的研究者手册或已经批准产品的产品特性总结),因为它们是与产品存在因果关系,且反应严重,如下所述
指令2001/20/EC第2(o)条对严重不良反应的定义:“任何不当的医疗事件或影响,在任意剂量下导致死亡、危及生命、需要住院或延长现有住院、导致持续或显著的残疾或丧失能力,或先天异常或先天缺陷。”
对于未列入本清单的其他重要医疗结果,在决定是否需要报告时,应进行专业的医学和科学判断。
临床试验申请的准备和审查过程,以及监管者和申办方的早期咨询机会
- 强烈建议开发者在提交申请前的早期阶段与监管机构进行更多沟通,特别是针对高风险试剂,以确保有足够时间适当考虑任何安全问题,同时不会对产品开发造成不必要的延误。也应考虑增加监管机构与研究伦理委员会之间的沟通方式。
对于属于我们职责范围内高风险类别的新药物实体首次人体临床试验,建议并强烈推荐申办方与监管机构进行"提交前会议(pre-submission meetings)",以便双方识别潜在问题。如开发者选择不与监管机构进行提交前讨论,应要求他们在拟提交高风险药物首次人体临床试验申请前六周通知监管机构。这将给予监管机构充分时间考虑是否需要外部专家意见,并确定合适的专家(见下一条建议)。提前通知可以减少延长评审过程的可能性。
- 对于高风险药物试验申请的评估,根据药物的性质、创新程度、预期药理靶点以及目标接受者,监管机构应该能够获得来自独立专业专家的额外意见,这些专家在其研究领域有丰富的知识。
- 人用药品委员会的专家咨询小组(EAG),或类似机构,可以承担这一角色,其核心成员由适当的专家组成,并能够根据需要吸收额外的专业知识。
- 在临床试验评估的时间安排上,对于一些异常复杂的特殊情况,应考虑引入一定的灵活性。
我们强调“异常复杂的特殊情况”;并无意在新药开发的监管过程中引入不必要的延误。《欧洲临床试验指令》(第6条第7款)(指令2001/20/EC,以下简称)已经允许对基因治疗、体细胞治疗以及含有转基因生物的产品的评估时间进行延长。这可能成为其他将新科学引入治疗的新型创新药物的参考模式。
确定并管理人体的起始剂量
- 对于那些其主要药理作用在动物模型中无法证明其拟议治疗效果的新药物,应给予特别关注。任何此类试验的剂量选择依据都应包括对拟议作用机制的明确理由,以及该物质在预期临床用途中的安全性和有效性的说明。
- 在剂量计算时,应采取比仅依赖动物研究中的“未观察到效应水平(NOEL, No Observable Effect Level)”或“未观察到不良效应水平(NOAEL, No Observable Adverse Effect Level)”更广泛的方法。起始剂量的计算应利用所有相关信息。需要考虑的因素包括药物的新颖性、生物活性及作用机制、药物的物种特异性程度、人类和动物细胞生物效应的剂量-反应曲线、体内动物实验的剂量-反应数据、药物代谢动力学和药物效应动力学建模、靶点占有率与浓度的计算以及人体体内靶点或靶细胞的暴露量的计算。
“最低预期生物效应水平(MABEL, minimum anticipated biological effect level)”方法是实现这一目标的一个很好的模型。(参见BIA/ABPI报告及利益相关者提交。)
- 如果不同的方法给出的人体安全剂量估计不同,首次人体试验的起始剂量应该取最低值,并在计算实际起始剂量时引入"安全冗余量(margin of safety)"。
- 当临床前信息出于任何原因,可能无法很好地指导人体体内反应时,首次人体试验的起始剂量应以谨慎为原则进行计算。之后的剂量增加也应小心进行,因为起始剂量可能特别低,而且剂量-反应曲线可能很陡峭。
- 在首次人体试验中,应该仔细考虑首个剂量的给药途径和速度,并密切监测是否出现"不良反应或过度反应(adverse or exaggerated response)"。
例如,对于首次静脉注射高风险药物的人体暴露,几小时的缓慢输注可能比几分钟的缓慢推注更合适。这可以让医生监控不良反应,并在临床上有必要时停止输注。
- 试验设计,包括受试者人数、起始剂量的决定以及剂量递增方案,应该根据具体情况来制定,并且在科学和统计上都有合理依据,同时考虑所有相关信息。
(参见英国皇家统计学会的相关意见)
- 在首次人体试验中,新药物应按顺序给不同受试者使用,并在每次给药之间留有适当的观察时间。受试者之间的观察间隔应根据药物的性质、靶点和受体特征,以及药物的潜在药代动力学、毒理动力学来预估可能出现的不良反应。
不同受试者之间以及同一受试者的重复给药之间都应留有合适的间隔,并且间隔时间都应基于现有证据加以合理说明。
- 在剂量递增的过程中,受试者之间连续给药时,也应进行类似的监测周期(如上所述)。
- 是否在健康志愿者或患者志愿者中进行首次人体试验,应经过仔细考虑并充分论证,同时考虑所有与受试者安全性相关的因素以及可能获得的科学信息价值。
一般来说,在新药的首次人体试验中,患者没有预期的直接获益。因此,风险与收益的评估通常不是决定是否应在健康志愿者或患者志愿者中进行此类试验的主要因素。
在癌症药物领域中可能存在例外情况,因为经历试验药物有效反应的患者通常能够继续使用该药。在这种情况下,风险与收益的平衡可能会成为决定由患者作为临床试验对象的因素,尤其是未对现有疗法有反应的患者,以及当试验药物具有可预测的细胞毒性(实际上可能是其预期的药理作用)时。
然而,最重要的因素应始终是志愿者的权利、安全和福祉,无论是患者还是健康个体,以及从临床试验中可以获得的信息价值。
首次人体试验的临床环境
- 负责首次人体试验受试者护理的主要研究者应始终具备适当的资格,并确保自己对试验药物、其作用靶点和作用机制有足够了解,从而能够做出知情的临床判断。
应大力鼓励为进行首次人体临床试验的主要研究者建立国家级职业认证体系。
- 在首次人体研究中,如果可预见会出现某些类型的严重不良反应,应提前考虑治疗策略。这应包括在有特定解毒剂可用时确保其可用性,并制定明确的支持性治疗计划,包括预先安排的重症监护(ITU)设施应急可用性。
- 对于高风险药物的首次人体试验,应始终在适当的临床环境中进行,由具备相应培训和专业知识的工作人员监督,并且应能立即使用用于处理和稳定急性紧急情况的设施,同时应事先安排好合理距离内的重症监护室(ICU)预案。
首次人体试验中应始终保证足够的工作人员配置,并在志愿者过夜时提供充足的24小时保障。
所有开展此类试验的临床地点都应有应急情况的标准操作程序,工作人员应通过定期演练保持执行这些程序的专业能力。试验参与者应始终被清楚告知在临床试验期间或之后出现不良反应症状时应该怎么做。
发展专业技能
- 应该扩大在临床试验的规划和执行中获得“实践”经验的机会。例如,研究生培训项目可以设有派遣到商业机构的交流期,或者在 NHS 和大学的专业中心进行培训的阶段(见下一条建议)。
英国医学科学院 2005 年发布的《更安全的药物》报告指出,需要培训新一代医生掌握药物安全性和有效性评估所需的技能。在最近 ABPI 进行的一项研究报告《维持制药和生物制药行业技能储备》中也发现了类似需求。
这需要高等教育资助机构和院校、NHS 以及行业之间的合作。鉴于主要利益相关方一致认同需要通过培养新一代临床药理学家及相关专业人员来填补这一技能缺口,这一问题应该可以得到解决,我们鼓励这样做,以确保未来首例人体临床试验的安全。
- 应探索建立针对高风险药物和先进药物产品的一期临床试验专科中心的可行性。
应鼓励开发国家级临床中心检查和认证系统,专门针对开展高风险药物人体首次试验的中心。认证应向所有符合规定标准的中心开放,包括公立和私立部门。
延伸阅读(Further reading)
‘Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Monoclonal Antibody TGN1412’
G. Suntharalingam, M. Perry, S. Ward, S. Brett, A. Castello-Cortes, M. Brunner, and N. Panoskaltsis.
N Engl J Med. 2006, 355:1018-1028.
The European Clinical Trials Directive 2001/20/EC (http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol- 1/dir_2001_20/dir_2001_20_en.pdf)
EudraLex Volume 10 –Clinical Trials (http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev10.htm)
European Union Guidelines - http://www.emea.europa.eu/htms/human/humanguidelines/background.htm
ICH E8 - General Considerations for Clinical Trials (CPMP/ICH/291/95) (http://www.emea.europa.eu/pdfs/human/ich/029195en.pdf)
ICH S6 Pre-clinical Safety Evaluation of Biotechnology-Derived Products (CPMP/ICH/302/95)
(http://www.emea.europa.eu/pdfs/human/ich/030295en.pdf)
ICH M3 (R1) Non-Clinical Safety Studies for the Conduct of Human Clinical Trials for Pharmaceuticals (CPMP/ICH/286/95). (http://www.emea.europa.eu/pdfs/human/ich/028695en.pdf).
ICH International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) http://www.ich.org/cache/compo/276-254-1.html
Academy of Medical Sciences 2005 report “Safer Medicines” (http://www.acmedsci.ac.uk/p102.html).
“Sustaining the skills pipeline in the pharmaceutical and biopharmaceutical industries”, ABPI. (http://www.abpi.org.uk/Details.asp?ProductID=285).
延伸阅读(翻译了文献标题)
‘在抗CD28单克隆抗体TGN1412一期临床试验中的细胞因子风暴’ G. Suntharalingam, M. Perry, S. Ward, S. Brett, A. Castello-Cortes, M. Brunner, 和 N. Panoskaltsis. 《新英格兰医学杂志》. 2006, 355:1018-1028.
欧洲临床试验指令 2001/20/EC (http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol-1/dir_2001_20/dir_2001_20_en.pdf)
EudraLex 第10卷 – 临床试验 (http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev10.htm)
欧盟指南 - http://www.emea.europa.eu/htms/human/humanguidelines/background.htm
ICH E8 - 临床试验的一般考虑 (CPMP/ICH/291/95) (http://www.emea.europa.eu/pdfs/human/ich/029195en.pdf)
ICH S6 生物技术衍生产品的临床前安全评估 (CPMP/ICH/302/95) (http://www.emea.europa.eu/pdfs/human/ich/030295en.pdf)
ICH M3 (R1) 药物开展人体临床试验的非临床安全研究 (CPMP/ICH/286/95) (http://www.emea.europa.eu/pdfs/human/ich/028695en.pdf)
ICH 国际人用药品注册技术要求协调会议 (ICH) http://www.ich.org/cache/compo/276-254-1.html
医学科学院 2005 报告 “更安全的药物” (http://www.acmedsci.ac.uk/p102.html)
“保持制药和生物制药行业技能储备”,ABPI. (http://www.abpi.org.uk/Details.asp?ProductID=285)